Глава 1.
Общие химические и экологические закономерности.
С чего начинается химия?
Cложный ли это вопрос? На него каждый ответит по-своему.
В середней школе учащиеся изучают химию в течение ряда лет. Многие довольно хорошо сдают выпускной экзамен по химии. Однако…
Беседы с абитуриентами и затем и студентами первых курсов говорят о том, что остаточные знания по химии после средней школы незначительные. Одни путаются в различных определениях и химических формулах, а другие вообще не могут воспроизвести даже основные понятия и законы химии, не говоря уже о понятиях и законах экологии.
У них химия так и не начиналась.
Химия, по-видимому, начинается с глубокого освоения ее основ, и прежде всего, основных понятий и законов.
1.1. Основные химические понятия.
В таблице Д.И.Менделеева рядом с символом элемента стоят цифры. Одна цифра обозначает порядковый номер элемента, а вторая атомную массу. Порядковый номер имеет свой физический смысл. О нем мы будем вести разговор позже, здесь остановимся на атомной массе и выделим в каких единицах она измеряется.
Следует сразу оговориться, что атомная масса элемента, приведенная в таблице, величина относительная. За единицу относительной величины атомной массы принята 1/12 часть массы атома углерода, изотопа с массовым числом 12, и назвали ее атомной единицей массы /а.е.м./. Следовательно, 1 а.е.м. равна 1/12 части массы изотопа углерода 12
С. И она равна 1,667*10–27
кг. /Абсолютная масса атома углерода равна 1,99*10–26
кг./
Атомная масса
, приведенная в таблице, является массой атома, выраженной в атомных единицах массы. Величина безразмерная. Конкретно для каждого элемента атомная масса показывает, во сколько раз масса данного атома больше или меньше 1/12 части массы атома углерода.
Аналогичное можно сказать и о молекулярной массе.
Молекулярная масса
– это масса молекулы, выраженная в атомных единицах массы. Величина тоже относительная. Молекулярная масса конкретного вещества равна сумме масс атомов всех элементов, входящих в состав молекулы.
Важным понятием химии является понятие «моль». Моль
– такое количество вещества, которое содержит 6,02*1023
структурных единиц /атомов, молекул, ионов, электронов и т.д./. Моль атомов, моль молекул, моль ионов и т.д.
Реклама

Масса одного моля данного вещества называется его молярной /или мольной/ массой. Она измеряется в г/моль или кг/моль и обозначается буквой «М». Например, молярная масса серной кислоты МН
2
SO4
=98г/моль.
Следующее понятие «Эквивалент». Эквивалентом
/Э/ называют такое весовое количество вещества, которое взаимодействует с одним молем атомов водорода или замещают такое его количество в химических реакциях. Следовательно, эквивалент водорода ЭН
равен единице. /ЭН
=1/. Эквивалент кислорода ЭО
равен восьми /ЭО
=8/.
Различают химический эквивалент элемента и химический эквивалент сложного вещества.
Эквивалент элемента – величина переменная. Она зависит от атомной массы /А/ и валентности /В/, которую элемент имеет в конкретном соединении. Э=А/В. Например, определим эквивалент серы в оксидах SO2
и SO3
. В SO2
ЭS
=32/4=8, а в SO3
ЭS
=32/6=5,33.
Молярную массу эквивалента, выраженную в граммах, называют эквивалентной массой. Следовательно, эквивалентная масса водорода МЭН
=1г/моль, эквивалентная масса кислорода МЭО
=8г/моль.
Химический эквивалент сложного вещества /кислоты, гидроксида, соли, оксида/– такое количество соответствующего вещества, которое взаимодействует с одним молем атомов водорода, т.е. с одним эквивалентом водорода или замещает такое количество водорода или любого другого вещества в химических реакциях.
Эквивалент кислоты
/ЭК
/ равен частному от деления молекулярной массы кислоты на число атомов водорода, участвующих в реакции. Для кислоты H2
SO4
, когда оба атома водорода вступают в реакцию H2
SO4
+2NaOH=Na2
SO+2H2
O эквивалент будет равен ЭН
2
SO4
= МН
2
SO
4
/nН
=98/2=49
Эквивалент гидроксида /Эгидр.
/ определяется как частное от деления молекулярной массы гидроксида на число гидроксогрупп, вступающих в реакцию. Например, эквивалент NaOH будет равен: ЭNaOH
=МNaOH
/nОН
=40/1=40.
Эквивалент соли
/Эсоли
/ можно рассчитать, поделив ее молекулярную массу на произведение числа атомов металла, вступающих в реакцию, и их валентность. Так, эквивалент соли Al2
(SO4
)3
будет равен ЭAl
2
(
SO
4
)
3
=МAl
2
(
SO
4
)
3
/6=342/2,3=342/6=57.
Эквивалент оксида
/Эок
/ можно определить, как сумму эквивалентов соответствующих элемента и кислорода. Например, эквивалент СО2
будет равен сумме эквивалентов углерода и кислорода: ЭСО
2
=ЭС
+ЭО
=3+8=7.
Реклама

Для газообразных веществ удобно пользоваться эквивалентными объемами /ЭV
/. Так как при нормальных условиях моль газа занимает объем 22,4л, то исходя из этой величины, легко определить эквивалентный объем любого газа. Рассмотрим водород. Мольная масса водорода 2г занимает объем 22,4л, тогда его эквивалентная масса 1г занимает объем 11,2л /или 11200мл /. Следовательно ЭV
Н
=11,2л. Эквивалентный объем хлора равен 11,2л /ЭVCl
=11,2л/. Эквивалентный объем СО равен 3,56 /ЭVC
О
=3,56л/.
Химический эквивалент элемента или сложного вещества используется в стехиометрических расчетах обменных реакций, а в соответствующих расчетах окислительно–восстановительных реакций применяют уже окислительный и восстановительный эквиваленты.
Окислительный эквивалент
определяют как частное от деления молекулярной массы окислителя на число электронов, которое он принимает в данной окислително–восстановительной реакции.
Восстановительный эквивалент равен молекулярной массе восстановителя поделенной на число электронов, которое он отдает в данной реакции.
Напишем окислително–восстановительную реакцию и определим эквивалент окислителя и восстановителя:
5N2
aS+2KMnO4
+8H2
SO4
=S+2MnSO4
+K2
SO4
+5Na2
SO4
+8H2
O
Окислителем в этой реакции является перманганат калия. Эквивалент окислителя будет равен массе KMnO4
деленной на число электронов, принятых окислителем в реакции (nе=5). ЭKMnO
4
=МKMnO
4
/nе=158/5=31,5. Молярная масса эквивалента окислителя KMnO4
в кислой среде равна 31,5г/моль.
Эквивалент восстановителя Na2
S будет: ЭNa
4
S
=МNa
4
S
/nе=78/2=39. Молярная масса эквивалента Na2
S равна 39г/моль.
В электрохимических процессах, в частности при электролизе веществ, пользуются электрохимическим эквивалентом. Электрохимический эквивалент определяют как частное от деления химического эквивалента вещества, выделяемого на электроде, на число Фарадея /F/. Электрохимический эквивалент более подробно будет рассмотрен в соответствующем параграфе курса.
Валентность
. При взаимодействии атомов между ними образуется химическая связь. Каждый атом может образовывать только определенное количество связей. Количество связей предопределяет такое уникальное свойство каждого элемента, которое называют валентностью. В наиболее общем виде валентностью называют способность атома образовывать химическую связь. За единицу валентности принимают одну химическую связь, которую способен образовать атом водорода. В связи с этим, водород является одновалентным элементом, а кислород – двухвалентным, т.к. с атомом кислорода могут образовывать связь не более двух водородов.
Умение определять валентность каждого элемента, в том числе и в химическом соединении, является необходимым условием успешного усвоения курса химии.
С валентностью соприкасается и такое понятие химии как степень окисления
. Подстепенью окисления понимают тот заряд, который имеет элемент в ионном соединении или имел бы в ковалентном соединении, если бы общая электронная пара бала бы полностью смещена к более электроотрицательному элементу. Степень окисления имеет не только цифровое выражение, но и соответствующий знак заряда (+) или (–). Валентность не имеет этих знаков. Например, в H2
SO4
степень окисления: водорода +1, кислорода –2, серы +6, а валентность, соответственно, будет 1, 2, 6.
Валентность и степень окисления в числовых значениях не всегда совпадают по величине. Например, в молекуле этилового спирта СН3
–СН2
–ОН валентность углерода 6, водорода 1, кислорода 2, а степень окисления, например, углерода первого –3, второго –1: –3
СН3
––1
СН2
–ОН.
1.2. Основные экологические понятия.
За последнее время понятие “экология” глубоко входит в наше сознание. Это понятие, введенное еще в 1869г Э.Геккелем /происходит от греческого oikos
– дом, место, жилище, logos
– учение/ все больше и больше тревожит человечество.
В учебниках биологии экологию
определяют как науку о взаимоотношениях живых организмов и среды их обитания. Практически созвучное определение экологии дает Б.Небел в своей книге «Наука об окружающей среде» – Экология – наука о различных аспектах взаимодействия организмов между собой и с окружающей средой. В других источниках можно встретить и более широкое толкование. Например, Экология – 1/. Наука, изучающая отношение организмов и их системных совокупностей и окружающей среды; 2/. Совокупность научных дисциплин, исследующих взаимоотношение системных биологических структур /от макромолекул до биосферы/ между собой и с окружающей средой; 3/. Дисциплина, изучающая общие законы функционирования экосистем различного иерархического уровня; 4/. Комплексная наука, исследующая среду обитания живых организмов; 5/. Исследование положения человека как вида в биосфере планеты, его связей с экологическими системами и воздействие на них; 6/. Наука о выживании в окружающей среде. /Н.А.Агиджанян, В.И.Торшик. Экология человека./. Однако под термином «экология» понимают не только экологию как науку, а само состояние окружающей среды и его влияние на человека, животный и растительный мир.
В экологии часто пользуются таким понятием как экосистема. Экосистема является основной функциональной единицей экологии. Экосистемой
называют совокупность растений, животных и других организмов, взаимосвязанных между собой и с окружающей их средой, связанных таким образом, что система сохраняет свою устойчивость неограниченно долго. Понятие применяется как к системам, которые включают совокупность организмов, так и к системам, в которые входит один организм. Каждая экосистема является составной компонентой биосферы. Биосфера
представляет собой тонкий слой вокруг планеты Земля, где взаимодействуют между собой воздух, вода и земля и где обитают живые организмы.
Экосистемы или их звенья, наиболее чувствительные к неблагоприятному воздействию антропогенных нагрузок, называют критическими. В критических звеньях природных экосистем аккумулируются загрязняющие вещества и создаются высокие нагрузки на биоту.
Биотой
называют совокупность всех организмов экосистемы. Это исторически сложившаяся совокупность растений и животных, объединенных общей областью распространения. На биоту оказывает постоянное влияние абиотическая среда.
Абиотическая среда
– это совокупность условий неорганической среды, воздействующих на живые организмы. Влияние абиотической среды на живое вещество происходит химическим путем – через химический состав атмосферы, почвы, природных вод, донных отложений и физическим /или климатическим/ путем через такие показатели климатических условий как температура, осадки, ветер, давление атмосферы, строение земной поверхности. Абиотическая среда может меняться в зависимости от степени антропогенного воздействия на окружающую среду, от антропогенной нагрузки.
Антропогенной нагрузкой
называют созданный человеком искусственно или возникающей в результате его деятельности комплекс источников и факторов воздействия на окружающую среду. Антропогенная нагрузка может заключатся в интенсивном использовании природных ресурсов /например, добыча полезных ископаемых, вырубка леса и т.д./, а так же в загрязнении природной среды /вода, воздух, почва/ путем выброса в атмосферу вредных, загрязняющих веществ, сброс сточных вод и т.д.
Загрязняющими веществами /плютантами/ являются всевозможные химические соединения, повышенное содержание которых в биосфере и ее компонентах вызывает негативную токсико-экологическую ситуацию. По агрегатному состоянию загрязняющие вещества делятся на три группы: газообразные, жидкие и твердые. В связи с этим, возникает необходимость изучения всех трех агрегатных состояний химических соединений. С другой стороны, загрязняющие вещества классифицируют по их химической природе и их воздействию на живые организмы. Естественно, особую опасность представляют загрязняющие вещества, оказывающие мутагенное влияние, результатом которого могут быть нарушения в системе воспроизводства потомства, и концерогенное, обуславливающее развитие злокачественных новообразований.
Поллютанты способны разрушить гомеостаз. Гомеостаз – поддержание на постоянном уровне жизненно важных констант живой системы: для внутренней среды высших животных это рН, ионный состав крови, температура, для биосферы – целостность генофонда и замкнутость биотического круговорота. В этом отношении большую роль играет соблюдение предельно допустимых концентраций /ПДК/. ПДК – это максимальное содержание загрязняющего химического вещества, не вызывающее прямого или косвенного негативного влияния на окружающую среду и здоровья человека, а также не приводящее к накоплению токсичных элементов в сельскохозяйственных культурах. Сейчас контроль за поступлением в природу поллютантов /загрязняющих веществ/ ведется постоянно. Этот контроль называется мониторингом
.
Необходимо также сказать, что в экологии используются и такие понятия, как экологическая ниша, толерантность, токсикант и др.
Совокупность всех факторов среды в ареале /ареал – область распространения любой систематической группы организмов – популяций, вида, семейства/, при которых возможно существование определенного вида названа экологической нишей
. С экологической нишей связано явление толерантности. Толерантность
– способность организмов относительно безболезненно выносить отклонение факторов среды жизни от оптимальных для него. Однако в природе ничего не остается без последствий. Особенно влечет за собой последствия воздействие на природу веществ-токсикантов. Токсикант
– вредное химическое вещество, вызывающее отравление живого организма. О токсичности отдельных поллютантов будет рассказано в соответствующих параграфах курса.
1.3. Основные законы химии и экологии. Химико–экологические закономерности.
К основным законам химии относят так называемые стехиометрические законы. Стехиометрия
устанавливает соответствие между количеством реагентов, вступающих в химическую реакцию и количеством продуктов, образующихся в результате реакции. Это соответствие осуществляется стехиометрическими коэффициентами, проставляемыми в уравнение реакции.
Первый закон, который рассматривается в курсе химии – закон сохранения массы и энергии
. Можно по-разному подходить к этому закону, например, разделить на два:закон сохранения массы и закон сохранения энергии, или толковать его более широко, как закон сохранения материи. В экологизированном курсе химии целесообразно рассматривать эти законы отдельно, а затем сделать общий эколого-химический вывод.
Закон сохранения массы говорит о том, что в результате химических превращений сумма масс веществ до реакции и сумма масс веществ после реакции одинакова. Общая масса сохраняется, если даже в результате химического процесса получается небольшое количество полезного продукта. Следовательно, основная масса веществ идет в отходы. И стоит задуматься, правильно ли выбран данный технологический процесс? Куда девать получаемые отходы? На наш взгляд, этот закон имеет глубокое эколого-философское значение. Прежде всего, насколько возможны безотходные технологии о которых много говорили не так давно. С другой стороны, какова ответственность авторов-разработчиков того или иного технологического процесса, выбрасываемого в отходы значительные массы «ненужных», иногда очень вредных веществ.
По закону сохранении энергии «Любая энергия не исчезает и не возникает, а только одни ее виды переходят в другие в эквивалентных количествах». В этом законе мы сознательно на первое место поставили сохранение энергии, так как следует еще провести дополнительные исследования превращения химической энергии в другие виды. Особенно, если химическая реакция протекает в условиях экосистем и с загрязняющими веществами.
К этим законам мы будем возвращаться в процессе изложения курса и будем развивать эколого-химические идеи, заложенные в них.
Закон постоянства состава
раньше считали вторым по значимости среди химических законов. Он утверждает, что «каждое чистое вещество имеет постоянный качественный и количественный состав независимо от способов получения». Отсюда следует, что вещества, полученные разными способами, но имеющие один и тот же качественный и количественный состав, должны обладать одинаковыми химическими свойствами. Однако здесь необходимо сделать два уточнения. Во-первых, на химические свойства влияет не только качественный и количественный состав соединения, но и структура молекулы /взаимное расположение атомов/. В связи с этим, одно и тоже соединение, полученное разными методами, может отличаться по химическим свойствам. Но это не значит, что разный состав вещества, просто в различном порядке соединены друг с другом атомы. Во-вторых, закон постоянства состава выполняется при условии, что химическое соединение всегда состоит из одних и тех же изотопов данного элемента.
Закон кратных отношений
применим к соединениям, образующимся из двух элементов. Если два элемента образуют друг с другом несколько химических соединений, то массы одного элемента, приходящиеся на одну и ту же массу другого элемента, относятся между собой как небольшие целые числа. Например, углерод и кислород образуют два оксида: СО и СО2
. В этих оксидах массы кислорода, приходящиеся на одну и туже массу углерода, относятся как 1:2.
Закон оъемных отношений
свидетельствует о том, что объемы взаимодействующих газообразных веществ относятся между собой и к объемам продуктов реакции, как небольшие целы числа. Например, 2NO+O2
=2NO2
; VNO
:VO
2
:VNO
2
=2:1:2
Закон Авогадро
, сформулированный в 1811 году А.Авогадро, имеет большое значение для химии и физики газообразных веществ. По этому закону «В равных объемах различных газов при одинаковых условиях содержится одинаковое число частиц /молекул, атомов, ионов/. Из этого закона вытекает следствие: «Моль любого газа при нормальных условиях занимает объем 22,4л». Химикам известна также и величина, которую называют числом Авогадра «N». Число Авогадро показывает, сколько молекул содержится в одном моле вещества. N=6,02*1023
.
Закон эквивалентов
определяет, в каких количествах взаимодействуют вещества между собой. По этому закону «Химические вещества взаимодействуют друг с другом в весовых или объемных количествах, пропорциональных их эквивалентам». Эквивалентное количество образуется из продуктов реакции. Например, 2Н2
+О2
=2Н2
О. Здесь с четырьмя эквивалентами водорода взаимодействует четыре эквивалента кислорода и образуется четыре эквивалента воды.
Математически закон эквивалентов записывают следующим образом
m1
/m2
=Э1
/Э2
, или m1
/Э1
=m2
/Э2
=mn
/Эn
Законы экологии
.
Конкретная наука не всегда способна объяснить все многообразие явлений природы. Специальные науки изучают только отдельные грани природного явления и не затрагивают их связь с другими явлениями или другими гранями того же явления, изучаемого уже другой наукой. В тоже время в природе все взаимосвязано. И существуют более общие, главенствующие над всеми частными законами и закономерностями. Даже если они еще и не осознаны человеком. Это касается прежде всего экологии. Обратимся, например, к такому универсальному закону природы, как закону вектора развития
, который гласит, что «Развитие однонаправлено, от старости к молодости. Историю человечества нельзя вернуть вспять».
В этом законе заложена очень глубокая экологическая мысль. Если произойдет глобальная экологическая катастрофа, то все живое, в том числе и человек, погибнет, ибо исторического возврата в универсальном законе природы не предусмотрено. Эту мысль мы будем развивать в последующих главах учебника.
Ряд экологических законов или закономерностей открыты не так давно русскими и зарубежными исследователями. Так, В.И.Вернандский открыл: закон физико-химического единства живого вещества, закон константности и закон биогенной миграции атомов.
Согласно закона физико-химического единства живого вещества
«Все живое вещество Земли физико-химически едино». А по закону константности
«Количество живого вещества биосферы /для данного геологического периода/ есть константа». Причем, по биогенной миграции атомов
«Миграция химических элементов на земной поверхности /и в биосфере в целом/ осуществляется или при непосредственном участии живого вещества или же она протекает в среде геохимические особенности которой обусловлены живым веществом /как тем, которое в настоящее время населяет биосферу, так и тем, которое действовало на Земле в течение всей геохимической истории/ ».
С законом вектора развития созвучен закон необратимости эволюции Л.Долло
«Организм /популяция, вид/ не может вернуться к прежнему состоянию, уже пройденному его предками». Что касается живых организмов, то по закону минимума Ю.Либиха
«Выносливость организма определяется самым слабым звеном в цепи его экологических потребностей, то есть жизненные возможности лимитирует тот экологический фактор, количество которого близко к необходимому организму или экосистеме минимуму и дальнейшее снижение которого ведет к гибели организма или деструкции экосистемы».
Важное значение имеет и закон максимума
, по которому «Количественное изменение экологических условий не может увеличить биологическую продуктивность экосистемы и хозяйственную производительность агросистемы сверх вещественно-энергетических лимитов, определяемых эволюционными свойствами биологических объектов и их сообществ». В связи с этим, введен закон снижения энергетической эффективности природопользования
. По этому закону «В ходе исторического развития при получении полезной продукции на ее единицу в среднем затрачивается все большее количество энергии». Действие этого закона мы уже стали ощущать.
Глубокое философское и практическое значение имеют так называемые «Законы» экологии Б.Коммонера:
Первый – «Все связано со всем».
Второй – «Все должно куда-то деваться».
Третий – «Природа «знает» лучше».
Четвертый – «Ничто не дается даром».
И если к этим постулатам Б.Коммонера добавить закон неустранимости отходов или побочных воздействий производства
, который гласит, что «В любом хозяйственном цикле образующиеся отходы и возникающие побочные эффекты неустранимы, они могут быть лишь переведены из одной формы в другую или перемещены в пространстве», то становится ясным к каким экологическим последствиям может привести безрассудное хозяйствование, экологическая профанация и бездумное антропогенное воздействие на экосистемы. Мы почему-то не хотим до конца осознать, что тератогены
/вещества, воздействие которых на организм приводит к аномалиям в его развитии, возникновением уродств/ могут вызывать быстроразвивающуюся цепь появления сплошных мутантов. Мы все надеемся на толерантность
организма /способность организма относительно безболезненно выносить отклонение факторов среды от оптимальных для него/, ибо согласно закона толерантности
В.Шелфорда «Лимитирующим фактором процветания отдельного организма или вида может быть как минимум, так и максимум экологического воздействия, диапазон между которыми определяет выносливость организма к данному фактору».
За последнее время опыт показал, что этот диапазон резко сужается.
Надо иметь в виду и закон максимизации энергии
. Он объясняет, какая экосистема имеет больше шансов на выживание. По этому закону «Выживает та система, которая наилучшим образом способствует поступлению энергии и использует максимальное ее количество наиболее эффективным способом».
Глава 2.
Строение атома.
На рубеже XIX–XX веков наука вплотную подошла к открытию строения материи. В этот период, метко названный революцией в естествознании, были сделаны выдающиеся открытия:
–открытие катодных лучей /1897 – Крукс/,
–фотоэлектрического эффекта /1887 – Герц/,
–рентгеновских лучей /1895 – Рентген/,
–явления радиоактивности /1896 – Беккерель/,
которые подтвердили ранее сделанные предсказания о сложной структуре атома. В результате было обнаружено, что в состав атома входят отрицательно заряженные частицы, которые были названы Джозефом Томсоном – английским физиком – электронами.
Экспериментальным путем в 1911 году Эрнестом Розерфордом было открыто ядро атома, несущее положительный заряд и занимающее ничтожно маленькую часть пространства внутри атома.
Первые теории строения атома были примитивными и не получили широкого распространения. Однако в истории становления модели атома почетное место занимают теории Розерфорда и Бора. Розерфорд предложил планетарную модель атома /1911/. Бор сформулировал квантовые постулаты, разработал модель строения атома водорода, вывел формулы для расчета радиусов и энергии квантовых орбит и формулы для определения спектральных линий /1913/.
2.1. Квантово–механическая модель атома.
Современная квантово-механическая теория строения атома складывалась постепенно. Делались новые открытия, совершенствовался математический аппарат и, соответственно, выкристаллизовывалась модель атома. Современная квантово-механическая теория гласит, что атом любого элемента имеет сложную структуру. Положительная часть атома /положительный заряд/ сосредоточена в ядре. Отрицательную часть составляют электроны, которые находятся в беспрерывном движении.
2.1.1. Строение ядра. Протонно–нейтронная теория.
Ядро атома, открытое в 1911 году Розерфордом, имеет сложную структуру. Основными частицами, входящими в состав любого ядра, являются протоны и нейтроны.
Протон
/обозначается ¦р/ – элементарная частица, входящая в состав ядер всех атомов и имеющая массу, равную массе ядра атома водорода /1,008 а.е.м./ и заряд по величине равный заряду электрона, но противоположный по знаку /+1/.
Нейтрон
/обозначается 1
0
n/ – элементарная частица, обладающая массой близкой к массе протона /1,00866 а.е.м./, но не несущая электрического заряда /электронейтральная/.
Теория строения ядра атома, предложенная в 1932 году нашими исследователями Иваненко и Гапоном и немецким ученым Гейзенбергом названа протонно–нейтронной теорией ядра
. Согласно этой теории:
–ядро атома состоит из нуклонов /так названы в сумме протоны и нейтроны/;
–суммарное число протонов в ядре /Np=S¦р/ обуславливает величину положительного заряда ядра /Zя/. От него зависит число электронов в электронейтральном атоме /Ne/ и порядковый номер в таблице Менделеева /Z/:
Z=Ne=Zя=Np;
–суммарное число нейтронов /Nn=S1
0
n/ ccуммарным числом протонов /Np/ дают величину массы ядра /А=Np+Nn/. Эту величину называют массовым числом /А/. Массовое число А равно целому числу, наиболее близкому по значению к атомной массе данного элемента Аэ;
–зная заряд ядра и массовое число можно определить количество протонов в ядре:
Nn=А–Z;
–структура ядра атома может быть выражена следующей формулой:
Zp+(A–Z)n
Например, структура ядра атома фтора /А=19, Z=9/ будет 9р+10n, т.е. в состав ядра атома фтора входит 9 протонов и 10 нейтронов. Так как заряд ядра /Z/ и массовое число /А/ являются количественной характеристикой атома любого элемента /Э/, то он ставятся в виде индексов возле символа данного элемента A
Z
Э, например для фтора 19
9
F или для серебра 108
47
Ag.
Элементы, ядра атомов которых содержат одно и то же число протонов но различное количество нейтронов, названы изотопами
, например, цинк /Z=30, A=64; 66; 67; 68;70/ имеет изотопы 64
30
Zn, 66
30
Zn, 67
30
Zn, 68
30
Zn, 70
30
Zn.
Атомы элементов, имеющие одинаковые массовые числа, но различные заряды ядер, названы изобарами
, например: 40
18
Ar, 40
19
K, 40
20
Cr.
Химическим элементом
называют вид атомов, обладающих одинаковым зарядом ядра.
Наряду с протонами и нейтронами в состав ядер атомов входят и другие элементарные частицы, например, мезон. /Мезоны в двести-триста раз тяжелее электрона/. Существует мнение, что мезоны обуславливают ядерные силы, которые приводят к образованию прочных и компактных ядер из протонов и нейтронов. Этот аспект рассматривается в курсе ядерной физики.
2.1.2. Двойственная природа электрона.
Электроны, как элементарные частицы, проявляют корпускулярно-волновой дуализм. Они являются частицами и проявляют волновые свойства.
Любая частица представляет собой сосредоточение вещества в малой части пространства. Следовательно, как частицы электроны обладают массой me
и зарядом е.
Масса электрона me
=9,11*10–28
г. /в 1837,11 раз меньше массы атома водорода/. Заряд электрона е=1,6*10–19
Кл/ или 4,8*10–10
эл.ст.ед./. Движение электрона как частицы должно характеризоваться, с одной стороны, траекторией, т.е. координатами и, с другой стороны, скоростью в данный момент времени.
Однако в движении электроны проявляют волновые свойства. Этот процесс происходит в объеме трехмерного пространства и развивается во времени, как периодический процесс. Характеристикой волны является длина волны, ее частота, скорость движения и амплитуда с определенным знаком. Следовательно, электронный поток характеризуется длиной волны l, которую можно оценить с помощью уравнения Луи де Бройля /1924г./:
l=h/mv
Здесь h–постоянная Планка /h=6,62*10–34
Дж/, m–масса электрона, v–скорость электрона.
Можно сказать, что уравнение де Бройля объединяет характеристику волнового процесса /l/ и корпускулярного движения /mv–импульс/. Волновая природа электронов подтверждена экспериментально полученной картиной интерференции и дифракции электронов.
Неопределенность в поведении электрона
.
Поскольку электрон обладает волновыми свойствами, то его движение не может быть описано определенной траекторией. Траектория «размывается», возникает область /полоса/ неопределенности, в пределах которой и находится электрон.
В связи с этим, для электрона, как микрочастицы, применим принцип /соотношение/ неопределенности Гейзенберга
/1927/, который гласит, что в любой момент времени невозможно одновременно точно определить и положение электрона в пространстве /его координату/ и его скорость /импульс/, минимальная возможная неточность равна h.
Математически принцип неопределенности можно выразить так:
(Dpx
)(Dx)=>h
Здесь Dpx
–неопределенность в величине импульса,
Dx – неопределенность в положении частицы в пространстве,
h – постоянная Планка.
Так как h– величина постоянная, то из принципа неопределенности следует, что чем точнее будем определять импульс электрона / его скорость /, тем большую будем допускать ошибку в определении его координаты, т.е. местонахождения.
В соответствии с принципом неопределенности траекторию электрона нельзя рассматривать со строгой математической точностью, как боровскую орбиту, существует область неопределенности, в которой может двигаться электрон. Поэтому следует говорить только о вероятности того, что электрон в данный момент времени будет в данном месте пространства атома.
В квантовой механике имеют дело со статическими принципами и вероятностным характером поведения электронов. Область пространства атома, внутри которой существует наибольшая вероятность нахождения электрона, называется орбиталью
.
2.1.3. Волновая функция и волновое уравнение.
Так как электронам присущи волновые свойства и они обладают неопределенностью положения в пространстве, их движение характеризуется при помощи волновой функции y и описывается волновым уравнением. Физический смысл волновой функции заключается в том, что ее квадрат y2
пропорционален вероятности нахождении электрона в элементарном объеме атома DV с координатами x, y, z.
Значение волновой функции находят при решении волнового уравнения Шредингера:
s2
y/sx2
+ s2
y/sy2
+ s2
y/sz2
+8p2m/h2
*(E–U)y=0
В этом сложном дифференциальном уравнении с частными производными: Е–полная энергия частицы, U – потенциальная энергия, y –волновая функция.
 Волновая функция, получаемая при решении уравнения Шредингера, может иметь ряд значений. Эти значения зависят от квантовых параметров n, l, me
, названных квантовыми числами Волновая функция, получаемая при решении уравнения Шредингера, может иметь ряд значений. Эти значения зависят от квантовых параметров n, l, me
, названных квантовыми числами
 n n
  yl yl
me
В итоге – значения квантовых чисел есть не что иное как результат решения уравнения Шредингера. Следовательно, при решении уравнения Шредингера получены значения волновой функции и возможные /допустимые/ значения квантовых чисел.
2.1.4. Квантовые числа. Атомные орбитали.
Так как электрон имеет четыре степени свободы, то для характеристики его поведения в атоме требуется четыре квантовых числа.
Главное квантовое число
n
определяет удаленность атомной орбитали от ядра и характеризует общий запас энергии электрона на данном энергетическом уровне. n принимает целочисленные значения от единицы до бесконечности. В зависимости от цифровых значений главного квантового числа приняты буквенные обозначения квантовых уровней n=1, 2, 3, 4,…
обозначение К, L, M, N,…
Чем больше n, тем слабее электрон связан с ядром и более емким становится квантовый уровень. Числовые значения nопределяют также и количество подуровней, содержащееся на данном квантовом уровне /т.е. числовые значения n определяют емкость квантового уровня/. Так, если n=3, то это значит, что имеем третий квантовый уровень, который состоит из трех подуровней.
Орбитальное квантовое число
l
характеризует момент количества движения электрона относительно центра орбитали. Наличие такого движения приводит к делению квантового уровня на подуровни. Орбитальное квантовое число характеризует так же пространственную форму электронного облака. Это квантовое число предопределяется главным квантовым числом n и принимает ряд целочисленных значений от нуля до n–1. В зависимости от числовых значений lприняты буквенные обозначения подуровней:
n=1, 2, 3, 4,…
l=0, 1, 2, 3,…,–1
обозначение подуровня: s, p, d, f,…
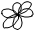     форма орбитали форма орбитали
Магнитное квантовое число
ml
характеризует магнитный момент электрона. Определяет ориентацию квантового подуровня в пространстве. Число проекций подуровня на направление магнитных силовых линий квантуется и оно равно количеству орбиталей на данном подуровне. Можно сказать, что магнитное квантовое число определяет количество орбиталей на подуровне. ml
принимает значения от –l через ноль до +l.
ml
= –l,…,+1, 0, –1,…, +l.
Рассмотрим подуровень s. Для него: l=0, ml
=0
  H рис.2.1. H рис.2.1.
 У подуровня шарообразной формы может быть только одна проекция. (рис.2.1.), имеющая значение «ноль». Следовательно, на s -подуровне только одна s-орбиталь. У подуровня шарообразной формы может быть только одна проекция. (рис.2.1.), имеющая значение «ноль». Следовательно, на s -подуровне только одна s-орбиталь.
 Подуровень Р имеет l=1, а ml
= –1, 0, +1 Подуровень Р имеет l=1, а ml
= –1, 0, +1
l=1
  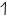      В данном случае согласно правил квантования уже три проекции. В данном случае согласно правил квантования уже три проекции.
  Следовательно на р-подуровне три р-орбитали. рис2.2. Следовательно на р-подуровне три р-орбитали. рис2.2.
Для d-подуровня: l=2, ml
= –2, –1, 0, +1, +2. Это значит, что согласно квантовой теории d-подуровень состоит из пяти d-орбиталей.
Подуровень f имеет l=3, ml
= –3, –2, –1, 0, +1, +2, +3. Следовательно f-подуровень состоит из семи f-орбиталей.
Число орбиталей на подуровне можно определить из выражения ml
=2l+1:
значение l: 0, 1, 2, 3, …….
подуровень: s, p, d, f, …….
число орбиталей: 1, 3, 5, 7, …….
Спиновое квантовое число
m
s
характеризует собственный момент количества движения, возникающий как бы из-за «вращения» электрона вокруг собственной оси. Принимает два значения: +1/2 и –1/2, что соответствует двум возможным направлениям вращения электрона.
ms
= +1/2; –1/2.
Оно получено из опытов Штерна и Герлаха.
Рассмотренные квантовые числа определяют энергию электрона, объем и форму пространства, в котором вероятно его пребывание в околоядерном объеме, т.е. размер, форму и ориентацию орбитали в пространстве.
Так как волновая функция y является решением уравнения Шредингера при всевозможных значениях квантовых чисел, то можно сказать, что волновая функция является в свою очередь функцией рассмотренных квантовых параметров n, l и ml
, где:
n= 1, 2, 3, 4,…,
l= 0, 1, 2, 3,…,n–1
ml
=–l,…, –1, 0, +1,…, +l
Атомные орбитали
. Так как вероятность нахождения электрона в пространстве далеком от ядра очень мала, когда говорят об орбиталях, то имеют в виду такую область вокруг ядра атома внутри которой сосредоточено 90–95% электронного заряда. С точки зрения квантовой механики атомные орбитали являются геометрическим изображением волновой функции y (n, l, ml
).
  ZЭлектронное облако
. Если бы в каждый момент времени ZЭлектронное облако
. Если бы в каждый момент времени
  y определяли положение электрона в трехмерном пространстве и y определяли положение электрона в трехмерном пространстве и
   ставили в том месте точку, то через множество таких определений ставили в том месте точку, то через множество таких определений
 X получили бы картину в виде пространственного облака изображен- X получили бы картину в виде пространственного облака изображен-
 ного точками с размытыми краями /рис.2.3.) ного точками с размытыми краями /рис.2.3.)
рис.2.3.
 Такое зарядовое облако называют электронным облаком. Его плотность, пропорциональная y2
, является непосредственной мерой вероятности нахождения электрона. Граничная поверхность облака, внутри которой содержится 90–95% электронного заряда, дает форму орбитали. Такое зарядовое облако называют электронным облаком. Его плотность, пропорциональная y2
, является непосредственной мерой вероятности нахождения электрона. Граничная поверхность облака, внутри которой содержится 90–95% электронного заряда, дает форму орбитали.
 Zs-орбиталь
. Она существует при l=0. Значение ml
тоже равно Zs-орбиталь
. Она существует при l=0. Значение ml
тоже равно
Y нулю. Имеем только одно значение ml
=0. Следовательно,
s-орбиталь имеет максимальную симметричность. У нее
X сферическая форма (рис.2.4.). В этом случае вероятность на–
хождения электрона в околоядерном пространстве определя–
рис.2.4. ется только радиусом-вектором и не зависит от угла координат.
y Радиальное распределение электронной плотности для 1s Радиальное распределение электронной плотности для 1s
электрона соответствует кривой с максимумом (рис.2.5.).
Максимум распространения вероятности находится на
0 r1
r,A0
расстоянии от ядра r1
, которые соответствует радиусу
рис.2.5. первой боровской орбиты.
р-орбиталь
. Существует при l=1. ml
= –1, 0, +1.
 Z р-орбиталь появляется на втором и всех последующих Z р-орбиталь появляется на втором и всех последующих
  Рz уровнях. Так как ml
имеет три значения, то на р-подуров- Рz уровнях. Так как ml
имеет три значения, то на р-подуров-
Y не каждого уровне может быть три р-орбитали. р-орбиталь
 имеет гонтелеобразную форму. Все три р-орбитали распо- имеет гонтелеобразную форму. Все три р-орбитали распо-
X лагаются в пространстве по направлению координатных
Px осей. Их называют соответственно рх
, рy
, рz
-орбитали
Py (рис.2.6.).
 Рис.2.6. Z Y Y Z Z Рис.2.6. Z Y Y Z Z
X X X X Y
dz2
dx2
y2
dxy dxz dyz
рис.2.7.
d-орбиталь
. Появляется при l=2 на третьем квантовом уровне. На d-подуровне может быть уже пять различных состояний электронов, поэтому на d-подуровне каждого квантового уровня содержится пять d-орбиталей. В этом случае ml
принимает пять значений:ml
= –2, –1, 0, +1, +2, d-орбитали имеют более сложную форму, чем р-орбитали, они либо в виде четырех лепестков либо в виде гантели с ободком (рис.2.7.).
f-орбиталь
. Появляется при значении l=3. f-орбитали могут быть только на четвертом и более отдаленных уровнях. Так как при l=3 ml
имеет 7 значений /–3, –2, –1, 0, +1, +2, +3/, то на f-подуровне может быть семь орбиталей. Форма f-орбиталей еще более сложная, чем у d-орбиталей. f-орбитали изображают в виде сложных шестилепестковых фигур.
Форма орбиталей и ее направленность играют существенную роль при образовании химических связей, т.к. эти два фактора определяют характер и степень перекрывания электронных облаков соединяющихся атомов.
2.1.5. Структура электронных оболочек атомов.
Полная электронно-энергитическая структура атомов предопределяется набором рассмотренных квантовых чисел. Главное квантовое число n определяет не только номер квантового уровня, но и указывает на число подуровней содержащихся в данном уровне. Например, при n=3, имеем третий квантовый уровень, который состоит из трех подуровней: s-, p-, d-подуровня. Чем дальше от ядра находится квантовый уровень, тем он более емкий, тем из большего числа подуровней он состоит. Число орбиталей на уровне можно определять по формуле кn
=n2
, а число орбиталей на подуровне, как уже указывалось, по формуле кl
=2l+1.
Рассмотрим теоретическую схему взаимного расположения квантовых уровней и подуровней. /Фрагмент для первых четырех уровней/. На четырех вертикальных линиях отложим значения квантовых чисел n, l, ml
иms
.(рис.2.8.) На первой вертикальной линии изобразим квантовые уровни соответственно значениям квантового числа n /см. рис.2.8.). Мы уже знаем, что чем больше числовое значение n, тем более емкий квантовый уровень. По этому на рисунке он сделан более длинным по высоте. На второй вертикальной линии, отнесенной к квантовому числу l показано деление квантовых уровней на подуровни. Первый квантовый уровень состоит только из одного подуровня /обозначенного как s-подуровень/. Второй квантовый уровень делится уже на два подуровня: s-подуровень и р-подуровень. Третий уровень делится на три подуровня /s, p и d/, а четвертый – на четыре подуровня /s, p, d и f/.

n=4
|
–
–
–
–
|
f |
4f |
4f14
|
| d |
4d |
4d10
|
| p |
4p |
4p6
|
| s |
4s |
4s2
|
n=3
|
| d |
3d |
| p |
3p |
| s |
3s |
| n=2 |
–– |
p |
2p |
2p6
|
| s |
2s |
2s2
|
| n=1 |
– |
s |
1s |
1s2
|
n l ml
ms
кванто- подуровни орбитали электроны
вый уро- на орбиталях
вень рис.2.8.
Третья вертикальная линия соответствует квантовому числу ml
. Здесь показано деление квантового подуровня на орбитали. Число орбиталей на подуровне числу значений, которые принимает магнитное квантовое число. s-подуровень состоит только из одной орбитали, поэтому на первом квантовом уровне имеется только одна орбиталь. р-подуровень состоит из трех орбиталей.
ml
= –1, 0, +1
d-подуровень содержит уже пять орбиталей.
ml
= –2, –1, 0, +1, +2
f-подуровень увеличил свою емкость до семи орбиталей
ml
= –3, –2, –1, 0, +1, +2 +3
Четвертая вертикальная линия отнесена к спиновому квантовому числу ms
. Забегая вперед отметим, что это квантовое число предопределяет возможное количество электронов на орбитале. По соответствующему постулату на орбитале может быть два электрона, но они должны иметь разные спины, т.е. разные значения ms
: +1/2 и –1/2. В связи с этим на четвертой вертикальной линии представлена максимальная заполняемость электронами квантового подуровня и уровня.
На s-подуровне – 2 электрона
На p-подуровне – 6 электрона
На d-подуровне – 10 электрона
На f-подуровне – 14 электрона
Максимальное число электронов на подуровне можно определить по формуле:
К=2(2l+1).
Теоретическая последовательность расположения квантовых уровней и подуровней выглядит так:
1s-2s-2p-3s-3p-3d-4s-4p-4d-4f-5s-5p-5d-5f-6s-6p-6d-6f-7s-7p-7d-7f-…
Однако при расщеплении квантовых уровней на подуровни приведенная теоретическая последовательность нарушается. Реальное расположение подуровней определяется правилом Клечковского
, согласно которого последовательность расположения подуровней определяется суммарным значением двух квантовых чисел n и l. В том случае, когда для двух и более подуровней n + l имеет одинаковое значение, то сначала идет тот подуровень, у которого меньшее значение n.
1s – 2s – 2p – 3s – 3p – 3d – 4s – 4p – 4d – 4f – 5s –5p – 5d – 5f
(n+l) 1 2 3 3 4 5 4 5 6 7 5 6 7 8
По правилу Клечковского фактическая последовательность расположения подуровней следующая:
1s-2s-2p-3s-3p-4s-3d-4p-5s-4d-5p-6s-5d-4f-5d2–5
-6p-7s-6d1
-
5f- 6d2–5
-7p.
Структура электронных оболочек атомов изображена на следующей схеме (рис2.9.):
 |
6p |
| 5d4
|
| 4f 14
|
| 5d1
|
| 6s |
5p
4p
|
| 4d |
| 5s |
| 3d |
| 4s |
3p
2p
|
| 3s |
| 2s |
| 1s |
Рис.2.9. |
2.1.6. Основные принципы распределения электронов в атоме
.
Рассмотренная электронная оболочка атома заполняется электронами в соответствии трем принципам: принципу наименьшей энергии, принципу Паули /правилу/ Гунда.
Принципу наименьшей
гласит, что электрон в атоме занимает тот свободный подуровень, на котором он будут иметь минимальное значение энергии. По другому, электрон остается на том подуровне, на котором обеспечивается наиболее прочная связь с ядром.
Последовательность заполнения подуровней соответствует приведенному выше фактическому расположению подуровней в структуре электронной оболочки атома:
1s-2s-2p-3s-3p-4s-3d –… и т.д. /см. выше/.
Принцип Паули
/запрет Паули/ говорит о том, что в атоме не может быть даже двух электронов с одинаковыми значениями четырех квантовых чисел.
Следствие. На орбитале может находится два электрона с различными спинами (т.е. с различными значениями спинового квантового числа: ms
= +1/2 и ms
= –1/2).
Третий принцип – это принцип или правило Гунда
/Хунда/. Он объясняет порядок заполнения электронами квантового подуровня. В пределах подуровня электроны распределяются так, что их суммарное квантовое число имело максимальное значение /сначала по одному электрону на орбиталь, а затем спаривание/. Правильным будет распределение, например, трех р-электронов таким образом:
В зависимости от того, какой подуровень заполняется последними электронами, различают s-, p-, d-, f-элементы.
s-элементами называют такие элементы, в атомах которых последние электроны занимают s-подуровень внешнего квантового уровня. /Например, натрий, магний, калий, кальций и др./.
р-элементами называют такие элементы, в атомах которых последние электроны занимают р-подуровень внешнего квантового уровня. /Например, углерод, кислород, хлор и др./.
d-подуровень, заполняемый последними электронами, относит элементы к d-элементам. /Например, d-элементами являются титан, хром, железо, медь, т.к. у этих элементов последние электроны занимают d-подуровень предпоследнего уровня/.
f-элементами называют такие элементы, в атомах которых последние электроны занимают f-подуровень второго от вне квантового уровня. /Например, празеодим, европий, эрбий и др./.
2.1.7. Изображение электронной структуры атомов при помощи электронных формул и квантовых ячеек
.
Электронную структуру любого атома изображают электронными формулами. В электронных формулах квантовый электронный уровень обозначают численным значением главного квантового числа n, подуровень записывают буквенным обозначением соответствующего подуровня, а число электронов на подуровне указывают степенью, стоящей у обозначения подуровня. Например, 3d5
обозначает, что на d-подуровне 3-го квантового уровня находится пять электронов. Электронная формула любого элемента состоит из полного набора таких фрагментов, как указано в примере. Так, электронная формула атома титана /№22/ имеет вид: 1s2
2s2
2p6
3s2
3p6
4s2
3d2
. Cумма всех степеней равна 22, это значит, что атом титана, имея заряд ядра +22, содержит на электронной оболочке 22 электрона, которые своим суммарным отрицательным зарядом (–22) компенсируют положительный заряд ядра атома, делая атом сложной электронейтральной системой.
Электронные формулы удобнее составлять после рассмотрения структуры периодической системы элементов Д.И.Менделеева. Забегая вперед и основываясь на знаниях, полученных в средней школе, представим периодическую систему элементов в виде электронных формул. (рис.2.10.) О написании электронных формул конкретного элемента, находящегося в периодической системе, вернемся позже и рассмотрим алгоритм по которому легко научиться писать электронную формулу любого элемента. Во-вторых, структуру электронной оболочки атома изображают при помощи квантовых ячеек.
  I 1s2 I 1s2
II 2s2
2p6
III 3s2
3p6
IV 4s2
3d10
4p6
V 5s2
4d10
5p6
VI 6s2
5d1
4f14
5d9
6p6
VII 7s2
6d1
5f14
6d9
7p6
рис.2.10.
  Квантовыми ячейками мы уже пользовались хотя и не вводили понятие “квантовая ячейка”. Квантовая ячейка – это не что иное как графическое изображение орбитали. Ее показывают клеточкой , а электроны на орбитали изображают стрелкой, стоящей в клеточке . Для s-подуровня отведена одна клеточка, так как s-подуровень имеет только одну орбиталь. Для р-подуровня выделено три ячейки ибо р-подуровень содержит три р-орбитали. d-подуровень изображают пятью ячейками, а f-подуровень – семью ячейками. Квантовыми ячейками мы уже пользовались хотя и не вводили понятие “квантовая ячейка”. Квантовая ячейка – это не что иное как графическое изображение орбитали. Ее показывают клеточкой , а электроны на орбитали изображают стрелкой, стоящей в клеточке . Для s-подуровня отведена одна клеточка, так как s-подуровень имеет только одну орбиталь. Для р-подуровня выделено три ячейки ибо р-подуровень содержит три р-орбитали. d-подуровень изображают пятью ячейками, а f-подуровень – семью ячейками.
Электронная структура атома титана, для которого мы уже писали электронную формулу, изображенная при помощи квантовых ячеек, выглядит так:
2.1.8. Об индивидуальности каждого химического элемента
.
«Удостоверением личности» химического элемента можно назвать его электронную формулу. Глядя на нее химик скажет очень многое об индивидуальности данного «химического персонажа».
Мы уже знаем, что универсальной характеристикой элемента является положительный заряд ядра атома, а если смотреть еще глубже, то число положительно заряженных элементарных частиц – протонов. Увеличение их количества приводит к скачкообразному изменению свойств. Начинает действовать универсальный закон природы – закон перехода количества в качество. Однако на изменение качественное показателей элемента влияет не только число протонов, но и число нейтронов в ядре. Как уже было отмечено, элементы, имеющие одинаковое число протонов в ядре но разное количество нейтронов, названы изотопами. У каждого элемента свое число изотопов: у одного – больше, у другого – меньше. Когда в таблице Д.И. Менделеева указывают атомную массу элемента дробным числом, то это не значит, что в ядре имеется дробное число элементарных частиц, в этом случае взята средняя атомная масса всех изотопов данного элемента с учетом их количественного содержания в природе. Но если в точных экспериментах будем пользоваться его такой «усредненной» атомной массой, то это будет не совсем корректно, ибо за «усреднением» теряет свою индивидуальность данный изотоп, особенно, если изотоп радиоактивный.
На практике еще больше отклоняются от истинного значения, когда пользуются не атомной массой, а массовым числом «А», т.е. целым число, самым близким к атомной массе. Может быть, при рассмотрении индивидуальных особенностей элемента лучше брать атомную массу того изотопа, которого в процентном соотношении в природе больше, или того, который самый устойчивый /или неустойчивый/, если речь идет о радиоактивных элементах.
Как видим, закон перехода количества в качество для химических элементов реализуется по двум направлениям: по протонному и по нейтронному. По протонному: появление в ядре очередного протона скачкообразно приводит к новому элементу, а по нейтронному: расширяет качество данного элемента вплоть до появления радиоактивности. Это видно на примере водорода. Если к ядру водорода добавляется протон, то это уже отрицает все качества водорода как элемента /данного индивидуума/ и переводит его в новый элемент – гелий /т.е. в новый индивидуум/. Добавление нейтрона не отрицает самого элемента водорода, а расширяет границы его качества, образуя изотоп водорода 2
1
Н /названный дейтерием 2
1
Д / и далее изотоп 3
1
Н /названный тритием 3
1
Т/. С увеличением числа нейтронов элемент приобретает дополнительные признаки, в данном случае – радиоактивность.
Проявление отдельными изотопами радиоактивных свойств сообщает таким элементам особую индивидуальность, можно сказать опасную индивидуальность, если рассматривать элементы с экологических позиций. В этом отношении необходимо иметь «специальную таблицу Менделеева», в которой были бы представлены свойства радиоактивных элементов и форма их зависимости от положения в данной таблице. Такая таблица была бы полезной при использовании радиоактивных элементов в качестве «меченых атомов», а так же для экологических аспектов. /Таблица будет представлена в соответствующем параграфе курса/.
При определении индивидуальности химического элемента необходимо прежде всего условиться, для какой цели эта характеристика будет применяться. Потому что одно дело атомарное состояние химического элемента, а другое – то реальное состояние простого вещества, в котором данный элемент находиться в обычных условиях, т.е. в его стандартном состоянии. Если в современной периодической системе Д.И.Менделеева находиться 104–105 элементов, то число простых веществ возрастает до величины 250. И у каждого простого вещества своя специфическая индивидуальность.
Химические свойства элемента, его «химическая индивидуальность» определяется тремя его характеристиками: размером атома, энергией ионизации и сродством к электрону. Но как оценить размеры атома? Какую величину брать за радиус атома? Толи расстояние от ядра до максимума электронной плотности /одно значение/, или расстояние от ядра до граничной поверхности, в которой содержится 95% электронного облака /это уже другое значение/, а может размеры атома определять как полу расстояние между центрами двух одинаковых атомов в простой молекуле или в кристаллической решетке. /Это уже третье значение/. Чтобы результат был корректным, для сравнения и для обоснования какой-либо закономерности всегда необходимо брать величины, полученные одним и тем же методом. Для оценки «химической» индивидуальности элементов в экосистемах надо иметь свои критерии. Эти критерии будут изложены в соответствующем курсе.
Глава 3.
Периодический закон и Периодическая система элементов.
В 1969 году ученый мир отметил юбилейную дату – 100-летие со дня открытия Периодического закона химических элементов. В статье, посвященной столетию этого закона академик И.В. Петрянов–Соколов писал: « История – сурова. Она придирчиво сортирует все, что найдено и создано человеком. Очень немногое она хранит в течение века. Удивительная и привычная простота и четкость менделеевской таблицы из школьного учебника наших дней скрывает теперь от нас ту непостижимую, гигантскую кропотливую работу по освоению и переработке всего, что было найдено и познано до Менделеева, которую пришлось выполнить ему, чтобы стала возможной и осуществимой гениальная интуитивная догадка о существовании в мире Закона периодичности свойств элементов».
В прошлом веке химия стала развиваться ускоренными темпами. Накопилось большое количество опытных данных. Возникла необходимость систематизации химических элементов. Многие ученые до Менделеева принимались за эту работу, но никто не смог открыть всеобщую связь элементов, создать стройную систему, отображающую закон развития материи. Ни одна предлагаемая «Система» не могла удовлетворить ученых.
Д.И. Менделеев приступая к работе, четко представил себе, какие трудности его ожидают и чем может закончиться его поиск «Системы»: либо успехом, либо неудачей, как всех его предшественников.
3.1. Три этапа работы Д.И. Менделеева над проблемой систематики химических элементов.
Работу Д.И. Менделеева над вопросами систематики химических элементов можно логически разделить на три этапа:
Открытие Периодического закона;
Построение Периодической системы элементов;
Логические выводы, сделанные на основе Закона и Периодической системы.
Хотя все эти этапы переплетаются друг с другом, но для правильной оценки научного подвига нашего соотечественника рассмотрим каждый из этапов отдельно.
Открытие периодического закона.
Главная заслуга Д.И. Менделеева состоит в том, что он открыл фундаментальный закон природы – Периодический закон (1869г.).
 До Менделеева ни один ученый не смог обнаружить универсальной закономерности в существовании многообразия химических элементов. Ни «триады» Деберейнера, ни «октавы» Ньюлендса, ни «таблица» Мейера не отражали фундаментальной закономерности и не могли объяснить как сходство, так и различия между отдельными элементами. До Менделеева ни один ученый не смог обнаружить универсальной закономерности в существовании многообразия химических элементов. Ни «триады» Деберейнера, ни «октавы» Ньюлендса, ни «таблица» Мейера не отражали фундаментальной закономерности и не могли объяснить как сходство, так и различия между отдельными элементами.
К моменту начала работы Д.И. Менделеева над систематикой элементов существовало всего 63 химических элемента. Расположив элементы в порядке возрастания атомных масс, Д.И. Менделеев после длительного и глубокого анализа их свойств обнаружил универсальную закономерность, выражавшуюся в периодической повторяемости свойств через определенные интервалы элементов.
Следует заметить и тот факт, что в то время у ряда элементов неправильно были определены атомные массы, а значит элементы не могли находиться на своих законных местах, но вопреки этому Менделеев обнаружил закономерность. Д.И. Менделеев установил, что свойства элементов зависят от атомных масс и форма зависимости – периодическая.
Менделеев сформулировал открытый им закон так: «Свойства простых тел, а также формы и свойства соединений элементов находятся в периодической зависимости от величин атомных масс элементов».
Построение периодической системы элементов.
Периодическая система является графическим изображением периодического закона, и является естественным продолжением и развитием Менделеевым идей в области систематизации химических элементов.
Создавая периодическую систему элементов, Менделеев руководствовался не только атомными массами элементов, но и всей совокупностью их физических и химических свойств.
Второй этап работы Д.И. Менделеева был наиболее сложным. Во-первых, атомные массы ряда элементов (Be, Zn, In, Th и др.) были неправильно определены. Во-вторых, еще не был открыт целый ряд элементов.
Со всеми трудностями Д.И. Менделеев успешно справился и система была создана. Он распределил элементы на группы сходных по свойствам элементов, исправил атомные массы и оставил места для неоткрытых элементов.
Логические выводы
, сделанные Менделеевым, заключаются в следующим:
1). должны существовать и должны быть открыты неизвестные в то время элементы ;
2). должны быть исправлены атомные массы ряда элементов;
3). переход от типичных металлов к типичным неметаллам не должен быть очень резким.
Для некоторых элементов Менделеев оставил в таблице ряд свободных мест. Так были оставлены клеточки для элементов №21 (скандия), №31 (галлия), №32 (германия), №43 (технеция), эти элементы в течение 15 лет были открыты.
Менделеев очень подробно описал физические и химические свойства некоторых элементов. После открытия элементов, их свойства, установленные опытным путем, с удивительной точностью совпадали с предсказанными Менделеевым.
Будучи убежденным в том, что периодический закон отражает объективную реальность, он незамедлительно исправил атомные массы некоторых элементов ( Бериллия с 13,5 на 9; Индия с 76,6 на 113; Урана со 120 на 240; Тория с 116 на 232).
3.2. Современная формулировка периодического закона.
Д.И. Менделеев прекрасно понимал, что открытый им периодический закон и составленная на его основе периодическая система элементов обладает внутренней способностью к развитию. Современная квантово-механическая теория строения атома подтвердила правильность менделеевских воззрений на периодичность свойств химических элементов. Сейчас установлено, что главной характеристикой атома любого элемента является не атомная масса, а величина положительного заряда его ядра. Заряд ядра является наиболее универсальной характеристикой атома. От величины заряда ядра зависит общее число электронов в атоме и его положение в периодической системе (номер элемента в периодической системе равен величине заряда ядра. Заряд ядра определяет число электронов). От заряда ядра зависят свойства элементов. В связи с этим внесены уточнения в формулировке периодического закона. Современная формулировка периодического закона следующая:
Свойства элементов, формы и свойства соединений элементов находятся в периодической зависимости от величины заряда ядер их атомов.
Эта формулировка периодического закона не противоречит формулировке, данной Менделеевым. Она базируется на новых данных, которые придают закону и периодической системе научную обоснованность и подтверждают их правильность. Современная формулировка закона – это новый этап развития периодического закона, открытого Д.И. Менделеевым. Она легко объясняет те незначительные ономалии, которые встречаются в таблице Д.И. Менделеева. (Например, аргон с атомной массой 39,948 стоит впереди калия, атомная масса которого меньше, 39,102; теллур с атомной массой 127,60 стоит впереди йода, атомная масса которого равна 126,90).
3.3. Структура современной периодической системы элементов.
Д.И. Менделеев постоянно совершенствовал структуру периодической системы элементов. В 1871г он представил второй вариант системы – так называемую короткую форму таблицы. В этом варианте уже четко были выявлены различные степени сродства между элементами. Элементы разделены на восемь групп, номер группы равен высшей валентности, которую может иметь элемент.
Современная периодическая система элементов в общих чертах напоминает последние варианты менделеевской таблицы.
Сейчас наибольшее распространение имеют две формы периодической системы элементов: короткопериодная (табл. 3.1.) и длиннопериодная (табл. 3.2.) 105 элементов, известных в настоящее время, расположены в таблице в порядке увеличения заряда ядер атомов. Заряд ядра определяет порядковый номер элемента в периодической системе. Ключом к разгадке периодичности свойств элементов является строение электронных оболочек атомов.
Современная периодическая система состоит из 7 периодов и 8 групп. Периодом
называют последовательный ряд элементов, в пределах которого происходит постепенный переход от ярко выраженных металлических к ярко выраженным неметаллическим свойствам.
Например, второй период начинается типичным щелочным металлом (литием) и заканчивается двумя элементами (фтором и неоном) яркими неметаллами. Номер периода указывает на число квантовых электронных уровней в атоме и он равен значению главного квантового числа (n). Период начинается s-элементами и заканчивается p-элементами. s-элементами называют такие элементы, в атомах которых последние электроны заполняют s-подуровень внешнего квантового уровня. p-элементами – такие элементы, в атомах которых в последнюю очередь электроны заполняют p-подуровень внешнего уровня. Максимально в каждом периоде может быть только два s-элемента (например, Li и Be во втором периоде) и не более шести p-элементов (например, B, C, N, O, F, Ne во втором периоде).
Первых четыре периода являются малыми периодами. Причем первый период содержит только два элемента (H, He). Структура электронной оболочки, определяемая квантовыми числами, разрешает атому иметь на первом квантовом уровне только один подуровень (s-подуровень) с одной s-орбиталью, а следовательно на одной s-орбитали может быть только два электрона с разными спинами.
Второй, третий и четвертый периоды содержат по 8 элементов (s-элементов два и p-элементов шесть). Четвертый и все последующие периоды – большие. В короткопериодной системе большие периоды сложены в два ряда, но в длиннопериодной таблице большие периоды составляют один ряд. В больших периодах между s-элементами и p-элементами вклиниваются d-элементы. Максимальное число d-элементов в больших периодах – десять. d-элементами называют такие элементы, в атомах которых последние электроны заполняют d-подуровень предпоследнего уровня. Например, в четвертом периоде за двумя s-элементами (K, Ca) следует 10 d-элементов (от Sc до Zn). У d-элементов 4-го периода последние электроны заполняют d-подуровень третьего квантового уровня, т.е. 3d-подуровень.
Шестой и седьмой периоды становятся еще длиннее. В этих периодах появляются f-элементы. f-элементами называют такие элементы, в атомах которых последние электроны заполняют f-подуровень третьего от вне уровня, т.е. предпредпоследнего уровня. Например f-элементы шестого периода заполняют последними электронами 4f-подуровень. Максимальное число f-элементов в периоде – четырнадцать. f-элементы идут вслед за первым d-элементов, разбивая при этом d-подуровень на две части. Так в шестом периоде после двух s-элементов (Cs и Ba) идет один d-элемент под №57 (La). Затем следует четырнадцать f-элементов (Cs–Lu) и только после них, с №72 гафния продолжается d-подуровень (Hf–Hg) и заканчивается шестой период шестью p-элементами (Tl–Rn). Аналогичная картина имеет место в седьмом периоде. В короткопериодной форме периодической системы f-элементы вынесены в отдельную строчку и располагаются под таблицей. (ряд лантаноидов и ряд актиноидов).
 Если внимательно рассмотреть длиннопериодную форму периодической системы, то можно заметить, что с увеличением номера периода число неметаллов в периоде уменьшается. Практически неметаллы образуют компактный «треугольник». Если внимательно рассмотреть длиннопериодную форму периодической системы, то можно заметить, что с увеличением номера периода число неметаллов в периоде уменьшается. Практически неметаллы образуют компактный «треугольник».
Периоды.
I –– –– –– –– –– –– H He
 II –– –– B C N O F Ne II –– –– B C N O F Ne
III –– –– –– Si P S Cl Ar
IV –– –– –– –– As Se Br Kr
V –– –– –– –– –– Te J Xe
VI –– –– –– –– –– –– AtRn
В этом «треугольнике» два неметалла (H и He) являются s-элементами, остальные неметаллы относятся к p-элементам. Диагональ, выделенная рамочкой, содержит элементы, разделяющие неметаллы от металлов. Некоторые ученые выделенные рамочкой элементы, т.е. элементы, лежащие на диагональной границе треугольника (B, Si, As, Te, At), иногда называют полуметаллами или полунеметаллами по причине их двойственных свойств.
Группой
называют вертикальный ряд, столбец, элементов для которых существует идентичность свойств, т.е. группа – это совокупность элементов сходных по своим химическим свойствам. Группа делится на подгруппы. Рассмотрим два типа деления группы на подгруппы. Каждое деление основывается на своих принципах. Первая форма деления группы на подгруппы известна еще из средней школы: каждая группа делится на главную и побочную подгруппы. В главную подгруппу каждой группы входят элементы больших и малых периодов, а в побочную – только элементы больших периодов (d-элементы). Так, во второй группе к главной подгруппе относятся шесть элементов (Be, Mg, Ca, Sr, Ba, Ra), а к побочной подгруппе всего три элемента (Zn, Cd, Hg). По второму типу деления каждая группа делится на три подгруппы: подгруппу типических элементов и две подгруппы полных электронных аналогов.
В подгруппу типических
элементов входят элементы малых периодов, элементов, для которых наиболее ярко выражены свойства, определяемые номером группы.
Полными электронными аналогами
называют элементы, в атомах которых содержится одинаковое число электронов на внешнем и предпоследнем квантовых уровнях. Так вторая группа делится на следующие три подгруппы:
1. Подгруппа типических элементов – Be, Mg.
2. Подгруппа полных электронных аналогов кальция (подгруппа кальция) – Ca, Sr, Ba, Ra.
3. Подгруппа полных электронных аналогов цинка (подгруппа цинка) – Zn, Cd, Hg.
Особняком стоит восьмая группа. Она включает в себя пять подгрупп:
1. Подгруппу типических элементов – He, Ne.
2. Подгруппу полных электронных аналогов криптона (подгруппа криптона) – Kr, Xe, Rn.
3. Подгруппу железа – Fe, Ru, Os.
4. Подгруппу кобальта – Ko, Rh, Ir.
5. Подгруппу никеля – Ni, Pd, Pt.
В длиннопериодной таблице элементы главных подгрупп каждой группы называют просто – элементы IA группы; элементы IIA – группы и т.д. Элементы побочных подгрупп называют элементами Б групп – элементы IB – группы, элементы IIB – группы. Например: во IIA группу входят элементы Be, Mg, Ca, Sr, Ba и Ro. AIIB группа содержит элементы Zn, Cd, Hg.
3.4. Обзор закономерностей, выражаемых периодической системой элементов.
Закономерность изменения основных характеристик атомов предопределяется рамками периодической системы элементов. Опираясь на периодический закон, периодическую систему элементов, знание электронной структуры атомов можно достаточно точно описать свойства простых и сложных веществ. Свойства элементов в простых и сложных веществах в общем случае определяется размером атома (его радиусом) и структурой электронной оболочки.
3.4.1. Закономерность изменения радиусов атомов.
Так как движение электрона в атоме не имеет строгой, боровской траектории, а носит волновой характер, то и размер атома не имеет строго определенной границы. За радиус атома обычно принимают теоретически рассчитанные положения максимума плотности внешнего электронного облака. Такие радиусы называют орбитальными. Практически используют эффективные радиусы, которые определены из строения молекул и кристаллов. Радиусы атомов являются одной из важных характеристик элементов, т.к. размеры атомов определяют ряд физико–химических показателей и химическую активность элементов. Изменение атомных радиусов элементов носит периодический характер. Рассмотрим, как меняют радиуса атомов в пределах одного периода и одной группы. Такое рассмотрение сделаем на группе элементов второго периода и главной подгруппы первой группы. На приведенном ниже рисунке показан характер (тенденция) изменения радиусов атомов элементов второго периода. Значения радиусов даны в ангстремах А0
. (А0
=10 –8
см).

Li Be B C N O F
 А0 А0
1,52 1,13 0,88 0,77 0,70 0,60 0,66
Na – 1,86
K – 2,31 Характер уменьшения
радиусов атомов.
Rb – 2,44
Cs –2,62
Fr – 2,71 рис. 3.1.
В периодах радиуса атомов по мере увеличения заряда ядра, т.е. от начала к концу периода, уменьшается. Хотя в атомах элементов, находящихся в одном периоде, содержится одно и тоже количество электронных квантовых уровней, но по мере увеличения числа электронов происходит уменьшение радиусов атомов от начала к концу периода. Этот факт отличается тем, что при увеличении заряда ядра и числа электронов усиливается кулоновское взаимодействие между электронной оболочкой и ядром ( F=z*e/r2
), которое приводит к сжатию атома. Так, в ряду элементов второго периода от Лития до Фтора радиусы атомов уменьшались примерно в 2,5 раза.
В группах сверху вниз радиусы атомов увеличиваются, т.к. с каждым новым периодом появляется еще один квантовый уровень, который начинает заполняться электронами.
На рисунке стрелкой указана только общая тенденция изменения радиусов. Но это не значит, что в указанном направлении имеется линейная зависимость. На следующем рисунке отражен характер изменения радиусов атомов для интервала 100 элементов. (рис. 3.2.).
В рядах d-элементов изменения радиусов менее значительны, чем у s- и p-элементов. У d-элементов идет заполнение электронами d-подуровня предвнешнего квантового уровня и поэтому величина сжатия атома в целом меньше, чем в случае увеличения числа электронов на внешнем уровне. В ряду d-элементов величина сжатия радиусов атомов составляет всего около 0,3А0
(d-сжатие).
В ряду f-элементов величина сжатия еще меньше. Дело в том, что у f-элементов идет заполнение f-подуровня предпредпоследнего уровня, и увеличения заряда ядра и числа электронов очень мало влияет на размеры атомов. Величина f-сжатия составляет всего 0,1А0
. Однако это незначительное изменение радиусов в ряду f-элементов влияет на свойства последующих элементов. И, естественно, сами f-элементы, имея очень близкие радиусы атомов, схожи по химическим свойствам.
Полные данные по радиусам атомов представлены в Периодической системе Д.И. Менделеева, дополненной Кембелом значениями радиусов атомов. (табл.3.3.). Радиусы атомов были определены рентгеноскопическим методом
3.4.2. Закономерность изменения энергии ионизации.
Химическую активность элемента можно оценить способностью его атома терять и приобретать электроны. Способность атома отдавать электроны количественно оценивается энергией ионизации.
Энергией ионизации
называется такое количество энергии, которое необходимо затратить для отрыва одного элемента от нейтрального атома.
Энергию ионизации обозначают буквой I и выражают в кДж/моль или ЭВ/атом.
 A+I=A+
+e A+I=A+
+e
Многоэлектронные атомы характеризуются несколькими энергиями ионизации: I1
, I2
, I3
,…, соответствующими отрыву первого, второго, третьего и т.д. электронов. При этом, всегда I1
< I2
< I3
< In
, т.к. с увеличением числа отрываемых электронов растет заряд образующегося положительного иона, который сильнее притягивает электроны. Для характеристики химической активности элемента обычно пользуются значением первой энергии ионизации I1
(будем обозначать ее просто I). энергия ионизации тесно связана с размерами атома. Характер изменения энергии ионизации по периодам и группам рассмотрим на примере элементов второго периода и главной подгруппы первой группы. Результаты приведены на следующем рисунке. Значения I дается в ЭВ/атом.
Li Be B C N O F Ne
 ЭВ/атом ЭВ/атом
5,4 9,1 8,3 11,3 14,5 13,6 17,4 21,6
Na – 1,86
K – 2,31 Направление увеличения
энергии ионизации.
Rb – 2,44
Cs – 2,62
Fr – 2,71 рис. 3.3.
В периодах слева направо энергия ионизации атомов увеличивается. В группах сверху вниз – наоборот, энергия ионизации уменьшается. Из рисунка видно, что направление увеличения энергии ионизации соответствует направлению уменьшения радиусов атомов. Следовательно, чем меньше радиус атома тем труднее отрывать электрон, тем больше затрачена энергия ионизации.
Однако (как это видно из рис.3.4.) характер изменения энергия ионизации не соответствует прямой линии, но имеет периодический характер. В пределах каждого периода наблюдается «местные» максимумы. Это связано с порядком заполнения электронами квантовых подуровней. Во втором периоде сначала электроны заполняют s-подуровень, поэтому при переходе от элемента с электроном ns1
(Li) к элементу с электроном ns2
(Be) энергия ионизации возрастает. Затем идет скачек вниз (уменьшение) обусловленный заполнением электронами p-подуровня, но далее энергия ионизации возрастает при переходе от элемента с np1
(B) к элементу с nр3
(С).
Обусловленное заполнением подуровня по правилу Гунда (т.е. по одному электрону на орбиталь). Затем снова скачек вниз (уменьшение I). Начинается заполнение вторыми электронами np-подуровня. И энергия ионизации снова возрастает (от кислорода к неону). Местные максимумы и минимумы на возрастающем участке кривой в пределах подуровня отражает явление вторичной периодичности. Максимумы соответствует элементам, у которых внешние подуровни заполнены полностью ns2
, np6
или наполовину np3
. Это свидетельствует о повышенной устойчивости таких конфигураций.
В группах (в подгруппах s- и p-элементы) сверху вниз энергия ионизации уменьшается. Это обусловлено увеличением радиусов атомов: чем больше размер атома, тем легче от него оторвать электрон, тем меньше значение энергии ионизации.
В подгруппах d-элементов, кроме подгруппы скандия, как правило, сверху вниз повышается. Например:
VI1
=6,74 ЭВ/атом.
Nb I1
=6,88 ЭВ/атом.
Ta I1
=7,88 ЭВ/атом.
Повышение энергии ионизации в подгруппах d-элементов вызвано эффектом проникновения электронов к ядру. Согласно квантовой теории внешние электроны проникают ближе к ядру под d-подуровень. Это приводит к повышению прочности связи внешних электронов с ядром.
Данные по значениям первой энергии ионизации для значительного числа атомов представлены в таблице…
3.4.3. Сродство к электрону и характер его изменения.
Способность атома присоединять электроны может быть количественно оценена энергией, которую обозначают понятием «сродство к электрону».
Сродством к электрону называют количество энергии E, которое выделяется в результате присоединения электрона к нейтральному атому и превращением его в отрицательно заряженный ион.
А+е=А–
+Е
Сродство к электрону выражается в тех же единицах, что и энергия ионизации (кДж/моль или ЭВ/атом). Однако экспериментально его определить труднее, чем энергию ионизации. Поэтому надежные значения Е получены лишь для небольшого числа элементов. По имеющимся данным можно сделать однозначный вывод о закономерности изменения сродства к электрону по периодам и группам.
Характер изменения сродства к электрону рассмотрим на примере элементов второго периода и главной подгруппы седьмой группы показан на рис.3.5.
 Li Be B C N O F Ne ЭВ/атом Li Be B C N O F Ne ЭВ/атом
0,57 -0,6 0,2 1,25 -0,1 1,47 3,6 -0,57
3,8 – Cl
Увеличение сродства к
электрону 3,5 – Br
3,3 – I
– At рис.3.5.
Из приведенного рисунка следует, что в периоде от начала к концу сродство к электрону увеличивается, а в группах увеличение идет в направлении снизу вверх. Можно сделать такой вывод: чем меньше радиус атома, тем легче к нему присоединяется электрон, тем больше высвобождается энергии и, следовательно, больше сродство к электрону. Однако монотонности в изменении сродства к электрону нет, как и не было ее в изменении энергии ионизации.
Для элементов VIIA группы, обладающих в своих периодах наименьшими радиусами, величина сродства к электрону наибольшая. Наименьшее значение сродства к электрону и даже отрицательное значение имеет место у элементов с электронными структурами s2
(Be, Mg, Ca), s2
p6
(Ne, Ar, Kr) и с наполовину заполненным p-подуровнем, т.е. структурой s2
p3
(N, P, As). Это служит дополнительным доказательством повышенной устойчивости указанных конфигураций.
Изменение сродства к электрону в ряду d-элементов покажем на примере d-элементов 4-го периода.
| Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
| Е ЭВ/атом
|
–0,40 |
0,15 |
0,65 |
0,85 |
–1,20 |
0,1 |
0,70 |
1,10 |
1,40 |
–0,9 |
| электронная конфигурация |
s2
d1
|
s2
d2
|
s2
d3
|
s1
d5
|
s2
d5
|
s2
d6
|
s2
d7
|
s2
d8
|
s1
d10
|
s2
d10
|
Здесь тоже устойчивые конфигурации s2
d5
, s2
d10
имеют отрицательное значение сродства к электрону. Принцип изменения сродства к электрону в ряде d-элементов такой же как у ряда s- и p-элементов.
Еще раз обратимся к характеру изменения Е в группах. Из данных, приведенных для галогенов, видно, что величина сродства к электрону у фтора (3,6) меньше, чем у хлора (3,8). Такую аномалию можно объяснить отталкиванием электрона в плотно заполненном 2р-подуровне. Такие аномалии встречаются у элементов других групп.
3.4.5. Изменение электроотрицательности.
Для того, чтобы решить вопрос: атом данного элемента легче теряет или присоединяет электрон – необходимо учесть энергию ионизации I и сродство к электрону Е. Сумму энергии ионизации и сродства к электрону называют электроотрицательностью (ЭО). Например, для нахождения электроотрицательности атома фтора (ЭОF
) необходимо суммировать его энергию ионизации (IF
) и сродство к электрону (ЕF
).
ЭОF
= IF
+ ЕF
=1736,36+339,74=2076,2 кДж/моль
Электроотрицательность измеряется в тех же единицах, что и энергия ионизации: кДж/моль или ЭВ/атом.
Однако для удобства применения вместо абсолютных значений электроотрицательности (кДж/моль или ЭВ/атом) используют значения относительной электроотрицательности (ОЭО или S). За единицу относительной электроотрицательности принята электроотрицательность атома лития.
ОЭО=ЭОLi
=ILi
+ELi
=S
Cоответственно определены величины ОЭО всех элементов. Их значения приведены в таблице 3.4.
Характер изменения относительной электроотрицательности элементов рассмотрен на примере элементов второго периода и главной подгруппы первой группы. (рис.2.6)
Можно сделать следующий вывод: чем меньше радиус атома, тем больше значение электроотрицательности.
Электроотрицательностью
называют свойство атома притягивать к себе электроны.
Наименьшим значением электроотрицательности обладают s-элементы, поэтому щелочные металлы легко отдают электроны. Их можно назвать наиболее электроположительными элементами. В противоположность щелочным металлам, галогены имеют большую электроотрицательность, поэтому они легко притягивают к себе электроны и с большим трудом отдают их.
Li Be B C N O F
1 1,5 2,0 2,5 3,0 3,5 4,0
Na – 0,97
K – 0,91 Увеличение S
Rb – 0,89
Cs –0,86
Fr – 0,7 рис. 3.6.
Наименьшим значением S обладает Fr(0,7) а наибольшим значением S обладает F(4,0). Понятие электроотрицательности служит также мерой относительной способности атомов в молекуле притягивать к себе электроны или оттягивать на себя электронную плотность.
По возрастающей величине относительной электроотрицательности неметаллы можно расположить в следующий ряд:
| Si |
At |
В |
Te |
P |
H |
As |
I |
S |
Sc |
C |
Br |
Cl |
N |
O |
F |
| 1,74 |
1,9 |
2,01 |
2,01 |
2,06 |
2,1 |
2,2 |
2,21 |
2,44 |
2,48 |
2,5 |
2,74 |
2,83 |
3,07 |
3,5 |
4,0 |
Как видно из приведенного каждый элемент в этом ряду обладает большей электроотрицательностью, чем элемент находящийся левее его.
3.4.6. Характер изменения валентности элементов.
Валентностью
называют свойство атомов данного элемента образовывать химические связи с атомами других элементов.
Валентность элементов обеспечивается так называемыми валентными электронами.
Валентными называются такие электроны данного элемента, которые образуют химические связи с атомами других элементов. Величина валентности атома данного элемента определяется числом неспаренных электронов, а также числом электронов, которые имеют возможность распариваться при незначительной затрате энергии.
У s-элементов валентными являются электроны s-подуровня внешнего квантового уровня. Например у атома магния с электронной структурой 1s2
2s2
2p6
3s2
3p0
валентными являются электроны 3s2
. Они могут распариваться с переходом одного электрона с 3s-подуровня на подуровень 3p. В возбужденном состоянии электронная структура магния будет 1s2
2s2
2p6
3s1
3p1
.
У р-элементов валентные электроны расположены на s- и p-подуровнях внешнего квантового уровня.
Так у атома алюминия с электронной структурой 1s2
2s2
2p6
3s2
3p1
валентными являются 3s2
3p1
-электроны. Причем один электрон с 3s-подуровня легко переходит на 3p-подуровень, образуется три неспаренных электрона. Поэтому алюминий –трехвалентный элемент.
У d-элементов валентными являются электроны расположенные на s-подуровне внешнего уровня и d-подуровне (предпоследнего) квантового уровня. Например, атом титана имеет электронную структуру 1s2
2s2
2p6
3s2
3p6
4s2
3d2
. Валентными для титана будут электроны 4s2
3d2
. В результате распаривания электронов 4s-подуровня получается четыре неспаренных электрона, которые и обеспечивают титану валентность четыре.
У f-элементов валентные электроны на s-подуровне внешнего уровня и f- подуровне предвнешнего (предпредпоследнего), т.е. третьего от вне квантового уровня.
Как правило, высшая валентность s- и p-элементов равна номеру группы, за исключением нескольких элементов второго периода (N, O, F). На примере s- и p-элементов третьего периода можно показать, что высшая валентность элемента равна номеру группы (табл.3.5.)
| Элемент |
Na |
Mg |
Al |
Si |
P |
S |
Cl |
| Номер группы |
I |
II |
III |
IV |
V |
VI |
VII |
Валентные
электроны
в невозбуж-
денном
состоянии
|
3s |
3s2
|
3s2
3p1
|
3s2
3p2
|
3s2
3p3
|
3s2
3p4
|
3s2
3p5
|
Расположение
валентных электронов в
возбужденном состоянии
|
3s1
|
3s1
3p1
|
3s1
3p2
|
3s1
3p3
|
3s1
3p3
3d1
|
3s1
3p3
3d2
|
3s1
3p3
3d3
|
| Высшая валентность |
I |
II |
III |
IV |
V |
VI |
VII |
Так как у элементов второго периода отсутствует d-подуровень, то азот, кислород и фтор не могут достигать валентности равной номеру группы. У них нет возможности распаривать электроны. У фтора максимальная валентность может быть равной единице, у кислорода два, а у азота – три.
Следует сделать здесь примечание. в данном случае разговор идет о главной (основной) валентности. Дальше будет показано, что наряду с основной валентностью атомы элементов способны проявлять и побочную валентность за счет образования дополнительных донорно-акцепторных связей.
Для большинства d-элементов высшая валентность может отличаться от номера группы. Валентные возможности d-элемента в конкретном, случае определяются структурой электронной оболочки атома. d-элементы могут иметь минимальную валентность выше номера группы (медь, серебро) и ниже номера группы (железо, кобальт). Например, серебро, находящееся в побочной подгруппе первой группы имеет соединения с валентностью III. Ag2
O3
, AgCl3
. Это выше номера группы. В тоже время кобальт в соединение проявляет валентность не выше III. (Co2
O3
), что ниже, чем номер группы (VIII).
С понятием валентность близко соприкасается второе понятие – степень окисления.
Степень окисления – это тот заряд, который атом имеет в ионном соединении или имел бы, если бы общая электронная пара полностью была бы смещена к более электроотрицательному элементу в ковалентном соединении. Следовательно, степень окисления в отличии от валентности характеризуется не только величиной, но и зарядом (+) или (–). Валентность имеет только величину и не имеет знака. Например, в сульфате натрия NaI
2
SVI
OII
4
валентность натрия, серы и кислорода равны соответственно I, VI, II. А степень окисления будет – натрия (+1), серы (+6), кислорода (–2). Валентность и степень окисления по величине не всегда совпадают. Так, в следующих соединениях CH4
, CH3
OH, HCOH, HCOOH, валентность углерода везде равна (IV), а степень окисления –4, –2, 0, +2 соответственно.
Для определения валентности элементов в соединениях следует использовать не только положением элемента в определенной группе в периодической системе, но и валентным так называемых эталонных элементов. К эталонным элементам относят такие, которые всегда имеют одинаковые значения валентности. Среди них:
Водород Н (I), Калий К (I)
Кислород О (II), Натрий Na(I)
Магний Mg (II), Алюминий Al(III)
Фтор F (I).
Что касается степени окисления то эти элементы могут служить эталонным для определения степени окисления других элементов в соединениях.
K+
, Na+
(+1), H+
(+1) (за исключением гидридов)
Mg+2
, Ca+2
(+2), F-1
(–1)
Al+3
(+3), Cl-1
(–1) (за исключением соединений с кислородом и фтором)
О-2
(–2) (за исключением соединений с фтором)
3.4.7. Характер изменения восстановительных и окислительных свойств элементов.
Если в химических реакциях элемент отдает электроны и повышает степень окисления, то он проявляет восстановительные свойства. Наоборот, в случае присоединения элементом электронов и понижении степени окисления, элемент проявляет окислительные свойства. Восстановительные и окислительные свойства элементов зависят от радиусов атомов. Чем меньше радиус атома, тем труднее элемент отдает электроны и слабее проявляет восстановительные свойства. В этом случае у элемента активнее будут проявляться окислительные свойства. В периодах слева направо восстановительные свойства элементов уменьшается, а окислительные – увеличиваются. В группах сверху вниз увеличиваются восстановительные свойства и уменьшаются окислительные.

Li Be B C N O F
 увеличение увеличение
Na окислительных свойств
элементов
K
Rb
Cs увеличение восстановительных свойств элементов
Fr рис. 3.7.
3.4.8. Характер изменения свойств однотипных соединений.
Поместим в ряд однотипные соединения галогенов – галогенводороды и рассмотрим, как изменяются их свойства (устойчивость соединений, степень диссоциации, сила кислоты, восстановительные свойства) в пределах главной подгруппы седьмой группы. Обнаруживается четкая закономерность, как и для простых элементов.
HF HCl HBr HJ
 возрастание радиуса галогена возрастание радиуса галогена
 увеличение увеличение
прочности
соединения увеличение степени
диссоциации
усиление кислотных
свойств
увеличение восстановительной активности галоген–иона
Так в направлении от фтора к йоду идет возрастание радиусов атомов, следовательно в этом направлении уменьшается прочность соединений. Чем больше радиус галогенов, тем менее прочно с ним связан водород. Сравним энергии Гиббса образования нескольких молекул.
D
G0
обр.
кДж/моль
|
HF |
HCl |
HBr |
| –273 |
–95 |
–53 |
Наиболее отрицательное значение энергии Гиббса образования имеет молекула HF, следовательно, она самая прочная. При переходе от HF к HBrD
G0
обр.
уменьшилось более, чем в пять раз. Соответственно, прочность молекул резко падает. Растворы галогенводородов в воде являются кислотами. При переходе от HF к HI увеличивается степень диссоциации кислоты, усиливаются кислотные свойства галогенводородной кислоты. (сила кислоты определяется концентрацией ионов Н+
, вернее ионов гидроксония Н3
О+
).
| кислота |
HF |
HCl |
HBr |
НI |
| степень диссоцации a, % |
7 |
78 |
89 |
90 |
В направлении от фтора к йоду увеличивается восстановительная способность галоген – иона. Так НI легко восстанавливает KMnO4
до двухвалентного состояния марганца (Mn2+
). Очень трудно справляется HCl, а HF вообще не в состоянии востановить марганец из перманганата калия.
Аналогичное можно продемонстрировать для однотипных соединений элементов шестой группы.
H2
O H2
S H2
Se H2
Te
 увеличение радиуса атома увеличение радиуса атома
увеличение силы кислоты
уменьшение прочности соединения
3.5. Закон Мозли.
Помещая в рентгеновскую трубку один за другим химические элементы и исследуя рентгеновские спектры этих элементов английский ученый Мозли в 1913г обнаружил, что с увеличением порядкового номера элемента одни и те же линии спектра смещаются в сторону уменьшения длин волн. При этом рентгеновское излучение не зависит от того, в каком виде находится данный элемент – в виде простого вещества или соединения.
Опираясь на экспериментальные данные Мозли установил, что частота колебаний рентгеновских лучей, испускаемых химическими элементами, линейно связана с порядковыми номерами атомов элементов. Мозли сформулировал следующий закон, который сейчас называется его именем:
Корни квадратные из обратных значений длин волн определенной линии характерестического рентгеновского спектра находится в линейной зависимости от порядковых номеров элементов.
Математически закон Мозли выражается следующей формулой:
 Ö1/x=a(Z–b) Ö1/x=a(Z–b)
Здесь х– выбранная длина волны рентгеновского спектра элемента; Z– порядковый номер элемента в периодической системе; а и b– коэффициенты: а– переменный коэффициент, который для каждой линии спектра имеет свое значение; b– постоянная экранирования или коэффициент заслона, указывающий на уменьшение величины заряда ядра к которому притягивается электрон (т.е. b– учитывает число квантовых электронных уровней).
Графически закон Мозли имеет следующий вид, изображенный на рис.3.8.

Ö1/x

 10 20 30 40 50 Z рис.3.8. 10 20 30 40 50 Z рис.3.8.
Na Ca Zn Zr Sn
Большое значение закона Мозли заключалось в том, что непосредственно из опыта можно было найти порядковые номера элементов и доказать, что они точно соответствуют номерам элементов в периодической системе Д.И. Менделеева.
Таблица атомных номеров Мозли составленная в 1914г, точно совпадала с таблицей Менделеева. В ней тоже были пустые места, как в таблице Менделеева, соответствующие неизвестным в то время элементам.
Для неизвестных элементов Мозли точно указал строение характеристических рентгеновских спектров, что привело к открытию в скором времени двух неизвестных
элементов: гафния (№72) и рения (№75).
Глава 4.
Химическая связь. Строение молекул.
Свойства химических соединений зависят от состава молекул, их строения и вида связи между атомами
Молекулой называют устойчивое образование (систему) из двух и более атомов.
Совокупность сил, удерживающих атомы в молекулах, называют химической связью. Прежде чем рассматривать характеристику химической связи, выясним природу сил, обуславливающую взаимодействие атомов и образование молекул.
4.1. Основные принципы взаимодействия атомов.
Взаимодействие между атомами происходит через поле. Основным полем является электрическое (электростатическое), т.к. гравитационное и магнитные поля в этом случае ничтожно малы. Рассмотрим частный случай: взаимодействие двух атомов водорода и образование молекулы водорода. Такое взаимодействие показано на рисунке 4.1. Когда атомы удалены друг от друга на большое расстояние, то силы, действующие между ними, равны нулю. При сближении двух атомов водорода между ними возникают два вида сил.

в е в
в а а
в
а
а е
НI
HII
Рис.4.1. Возникновение сил притяжения «а» и сил отталки–
вания «в» при сближении двух атомов водорода.
Между электронной оболочкой первого водорода НI
и ядром второго водорода HII
, так же и между ядром HI
и оболочкой HII
возникает сила притяжения. Их обозначим буквой а
. В результате притяжения двух атомов водорода энергия между ними (энергия системы из двух атомов) начинает уменьшаться. На энергетической диаграмме (рис.4.2.) это уменьшение энергии показано кривой “а
”. Но наряду с притяжением между двумя атомами водорода возникают силы отталкивания, обусловленные отталкиванием как между одноименно заряженными ядрами обоих атомов водорода, так и между электронными оболочками. Эти силы отталкивания на рис.3.1. показаны стрелками и обозначены буквой “в
”. В результате сил отталкивания энергия системы Н2
будет возрастать. На рисунке 4.2. увеличение энергии системы, вызванное силами отталкивания показано кривой “в
”.
Для получения реальной обобществленной картины изменение энергии системы необходима силы притяжения “а
” и силы отталкивания “в
”, т.е. сложить кривую “а
” и кривую “в
”.
При складывании сил “а
” с силами “в
” получаем результирующую силу “р
”.
 р=а+в р=а+в
На графике (рис.4.2.) результирующая сила даст новую кривую “р
”, показывающую характер изменения энергии системы Н2
в процессе сближения двух атомов водорода. На этой кривой “р
” имеется впадина(r0
) которую обычно называют потенциальной ямой. После точки r0
кривая “р
” резко идет вверх, т.е. энергия системы начинает стремительно возрастать. Притяжение двух атомов водорода заканчивается, когда между ними имеется расстояние rс
, соответствующее минимальному значению энергии.
 Е Е
в
НI
HII
HII
E0
r
DE p
a
Emin r0
rc
Рис.4.2. Изменение потенциальной энергии системы Н2
при сближении
двух атомов.
Расстояние rc
, является расстоянием между двумя центрами двух атомов в молекуле, называют длинной связи.
Из рисунка 4.2. видно, что расстояние меньше rc
, чем сумма двух радиусов атомов водорода 2R.

rc
<2R
R R
2Rrc
Рис.4.3.
Cледовательно, в системе двух атомов водорода, т.е. в молекуле водорода произошло перекрывание электронных оболочек взаимодействующих атомов.
Минимальному расстоянию между ядрами атомов водорода в молекуле Н2
, т.е. значению rc
, соответствует и минимальное значение энергии Emin. В процессе сближения двух атомов водорода происходит уменьшение энергии системы на величину DE. Эта величина может считаться энергией химической связи DE=Есв, т.к. для разрушения молекулы водорода на отдельные атомы необходимо затратить такое же количество энергии.
Из рассмотренного видно, что химическая связь основана на электрическом взаимодействии атомов. Следовательно, природа химической связи носит электростатический характер. В том случае, когда в результате образования молекулы, происходит перекрывание электронных оболочек взаимодействующих атомов (молекула Н2
и др.) на электрические силы химической связи накладываются силы квантового характера, обусловленные увеличением электронной плотности в области перекрывания электронных оболочек взаимодействующих атомов.
Основными характеристиками связи являются: энергия связи (Есв) и длина связи(rc
).
Для многоэлектронных атомов и при взаимодействии не двух, а более атомов кривая “р
” не всегда имеет такой вид, как для одноэлектронных атомов. В этом случае энергетические кривые как-то изменяют свои очертания, но ход их сохраняет типичные признаки: остаются нисходящие и восходящие ветви, а так же точки устойчивого равновесия с минимумом потенциальной энергии.
Так как при химических реакциях, а взаимодействие атомов и образование молекулы тоже химическая реакция, заряд ядра атома не меняется и устойчивыми остаются внутриэлектронные уровни, то химическая связь образуется, как правило, посредством так называемых валентных электронов – электронов, принимающих участие в образовании связи.
Из сказанного видно, что природа химической связи едина, имеет электрическое происхождение. Однако сама химическая связь в зависимости от характера соединения атомов друг с другом, … валентных электронов в веществе бывает различных типов.
4.2. Типы химической связи.
Все виды химической связи можно разделить на три группы связей:
–Валентные связи
–Дополнительные
–Металлические.
К валентным связям
относят ковалентную и ионную. Эти связи определяют валентность атомов в соединениях. В группу дополнительных связей входят донорно-акцепторная и водородная связи.
Эти типы связей приводят к образованию более сложных молекул из простых или к укреплению прочности молекулы за счет образования дополнительных внутремолекулярных связей без изменения валентности атомов.
Металлическая связь стоит особняком, она имеет место в твердых металлах и их сплавах.
Все типы связей можно объединить в такую диаграмму:
 Ковалентная Ковалентная
Валентные
Химическая связи Ионная
связь
Межмолекулярные Донорно-
Дополнительные акцепторная
связи
Внутримолекулярные Водородная
Металлическая
связь
4.3. Характеристика ковалентной связи.
В том случае, когда при взаимодействии двух атомов происходит перекрывание электронных оболочек (как это имеет место в молекуле водорода) химическая связь обеспечивается суммарными электрическими и квантовыми силами. Такая химическая связь называется ковалентной.
Для описания ковалентной связи применяют два метода: метод валентных связей (метод В.С.) и метод молекулярных орбиталей (метод М.О.). Каждый из этих методов дополняет друг друга. Если метод ВС хорошо объясняет полярность связи, насыщаемость, направленность геометрическую конфигурацию молекул, то метод МО более полно объясняет такие особенности молекул, как их магнитные свойства, спектральные характеристики и др.
4.3.1. Метод валентных связей.
Этот метод базируется на двух идеях:
1). Химическая ковалентная связь возникает в результате перекрывания валентных орбиталей взаимодействующих атомов. Каждая область перекрывания дает одну ковалентную связь. Получающаяся связь является двухцентровой , так как обобществленная электронная пара обслуживает центры (ядра) обоих атомов.
2). Молекула, образованная посредством такой связи, представляет собой конфигурацию, состоящую из двух практически неизменных фиксированных остовов. Под атомным остовом подразумевают ту основную часть атома, которая не принимает участия в образовании связи (т.е. атом без валентных электронов).
Полярность связи.
Место валентных связей хорошо и наглядно демонстрирует полярность ковалентной связи. Неполярная
ковалентная связь получается тогда, когда область перекрывания валентных орбиталей находится на одинаковом расстоянии от центров взаимодействующих атомов. Примером может служить связь в молекулах Н2
и Cl2
.

H2
Cl2
a б а б рис.4.4.
В обеих молекулах область перекрывания двух s-орбиталей (H2
) и двух р-орбиталей (Cl2
) находится на одинаковых расстояниях от центров двух атомов водорода и атомов хлора (т.е. а=б)
Графически молекулы с неполярной ковалентной связю обозначаются кружочком, в котором положительная и отрицательная полоса совпадают. (рис.4.5.)
 Рис.4.5. Рис.4.5.
Такая связь образовывается между атомами одного и того же элемента (водорода, кислорода, азота) или атомами различных элементов но с одинаковыми значениями электроотрицательности. Например, связь между бором и теллуром практически неполярна, т.к. у обоих элементов одинаковое значение относительной электроотрицательности (2.01)
Полярная ковалентная связь
образуется между атомами с различной электроотрицательностью. В этом случае область перекрывания взаимодействующих атомов смещается в сторону атома с большим значением электроотрицательности.
Такой, например, является связь между атомами в молекулах HCl, CO. В молекуле H–Cl (рис.4.6.) электоротрицательность элементов разная (Ен=2,1; Еcl=2,83)

H : Cl ; а>б
Рис.4.6.
а б
т.к. электоротрицательность хлора больше, то область перекрывания смещена в сторону. Аналогично в молекуле СО (Ес=2,5; Ео=3,5) область перекрывания смещена в сторону более электоротрицательного кислорода.
В молекулах с полярной ковалентной связью центры положительных и отрицательных зарядов не совпадают, в молекуле образуется два полюса: положительный и отрицательный. Такие молекулы называют дипольными (диполями).
Диполем называют систему, из двух равных по величине, но противоположных по знаку электрических зарядов (q) расположенных на некотором расстоянии друг от друга, называемом длиной диполя (l).
Графически дипольная молекула изображается так, как показано на рис.4.7.

q+
q–
l
Рис.4.7. Схема дипольной молекулы
 Степень полярности дипольных молекул характеризуется дипольным момен- том m. m=ql Степень полярности дипольных молекул характеризуется дипольным момен- том m. m=ql
Сделаем оценку величины дипольного момента полярной ковалентной связи. Рассмотрим такой крайний случай. Если бы область перекрывания (общая электронная пара) полностью сместилась к более электоротрицательному элементу, то заряд диполя q стал бы равен величине заряда электрона q=e=4,8*10–10
ел.ст.ед. (1,6*10–19
Кл) При средней длине диполя l=10–8
см величина дипольного момента следующая:
m=q*l= 4,8*10–10
* 10–8
ел.ст.ед.*см = 4,8*10–18
ел.ст.ед.*см
Величину 4,8*10–18
ел.ст.ед.*см принимают за единицу измерения дипольных моментов, названную Дебаем. (Д)
1Д = 4,8*10–18
ел.ст.ед.*см
В системе единиц СИ 1Д=0,33*10–29
К * м. Следовательно в нашем случае m=4,8Д.
Для полярной ковалентной связи величина дипольного момента лежит в интервале:
0< m < 4,8Д.
Например, дипольный момент молекулы СО равен 2,7Д (mсо
=2,7Д).
В случае полярной ковалентной связи, смещение электронной плотности к более электоротрицательному элементу, оба атома приобретают частичные заряды, обычно называемые эффективными. Например, в молекуле HF эффективный заряд у водорода равен +0,43, а у фтора –0,43.(H+0,43
–F–0,43
). В молекуле иодида водорода эффективные заряды значительно меньше (H+0,05
–J–0,05
). Следовательно, связь в молекуле HJ близка к неполярной. Действительно для этой пары атомов DE=2,21–2,1=0,11.
Значение дипольных моментов некоторых полярных молекул в дебаях:
HJ – 0,38, HCl – 1,03, NH3
– 1,57, H2
O – 1,84, HCN – 2,93.
Чем больше дипольный момент, тем тем сильнее выражена полярность молекулы. Дипольный момент имеет направление, а поэтому для сложных молекул дипольный момент складывается как векторная сумма отдельных связей. Так, как в молекуле H2
O связи находятся под углом
О
 Н Н Н Н
Полярность каждой связи О–Н составляет 1,51Д. Дипольный момент молекулы H2
O: mН
2
О
=1,84Д (векторная сумма двух связей ОН).
  Возможны такие случаи, когда отдельные связи в молекуле полярные, а дипольный момент молекулы равен нулю, например, в СО2
. Молекула СО2
линейна О=С=О. Каждая связь СО полярна (2,7Д), но дипольные моменты связей направлены в противоположные стороны (О=С=О) и суммарный дипольный момент равен нулю. Возможны такие случаи, когда отдельные связи в молекуле полярные, а дипольный момент молекулы равен нулю, например, в СО2
. Молекула СО2
линейна О=С=О. Каждая связь СО полярна (2,7Д), но дипольные моменты связей направлены в противоположные стороны (О=С=О) и суммарный дипольный момент равен нулю.
Постоянные дипольные моменты молекул имеют значения от нуля до 10Д. У неполярных молекул нет постоянного дипольного момента (m=0, так как l=0), у полярных m>0 и достигает 3,5–4Д. Дипольный момент ионных молекул достигает 10Д.
s- и
p–связи. Насыщаемость связи.
В зависимости от способа перекрывания валентных орбиталей взаимодействую-щих атомов различают s–и p– ковалентные связи.
 s–связью
называют такую ковалентную связь, для которой область перекрывания находится на линии связывающей центры взаимодействующих атомов, например, в молекулах H2
, HCl (рис.4.8.) s–связью
называют такую ковалентную связь, для которой область перекрывания находится на линии связывающей центры взаимодействующих атомов, например, в молекулах H2
, HCl (рис.4.8.)
Н Н HCl
а) б)
х х
ss
-
s
–cвязь ss
–р
–cвязь
Рис.4.8. а) молекулаH2
б) молекулаHCl.
В обоих случаях область перекрывания лежит на линии “х”, проходящей через центры атомов.
p связь
– это такая ковалентная связь, для которой область перекрывания располагается в плоскости перпендикулярной линии, связывающей центры взаимо-действующих атомов.
Рассмотрим молекулу кислорода О2
. Каждый атом кислорода имеет на внешнем квантовом уровне два неспаренных электрона, занимающих р-орбитали, например, рх
– и рy
–орбитали (рис.4.9.).
При взаимодействии друг с другом двух атомов кислорода происходит перекрывание попарно двух рх
–орбиталей и двух рy
–орбиталей. Причем, область перекрывания рх
–орбиталей находится на линии, связывающей центры двух атомов. При этом образуется spx
-
px
–связь. Область перекрывания двух рy
–орбиталей лежит в плоскости (x–y), т.е. в плоскости перпендикулярной линии связывающей центры кислородов. Образуется ppy
-
py
–связь. Это показано на рис.4.10.
 2 2 |
| n=1 |
p |
| s |
Pz
Py
Px spx-px
ppy
-
py
–связь
Рис.4.9. Рис.4.10. Образование s- и p–связей в молекуле О2
.
В молекуле N2
образуется две p–связи. Наряду с spx
-
px
–связью и ppy
-
py
–связью образуется вторая ppz
-
pz
–связь. Эта связь образуется в результате перекрывания pz
–орбиталей обеих атомов азота имеющих тоже по одному неспаренному электрону с противоположными спинами.
 ppz
-
pz ppz
-
pz
Nspx
-
px
N
ppy
-
py
p–связь вторичная после s–связи. Она образуется в том случае, когда уже имеется s–связь. Отдельно p–связь между двумя атомами не существует. p–связь как дополнительная менее прочная, чем s–связь. Возможность образования p–связи обеспечивает насыщаемость ковалентной связи и приводит к тому, что между двумя атомами могут быть не только одинарные, но и двойные и тройные связи.
Гибридизация связи. Направленность связи. Геометрическая конфигурация молекул.
Валентными являются электроны не только одного подуровня, орбитали которых имеют одинаковую форму, а и разных подуровней с различной конфигурацией электронных облаков. Например, атомы бария и углерода имеют валентные электроны, находящиеся на 2s и 2p-подуровнях (бор 2s2
2p1
; углерод 2s2
2p2
). В образовании связи принимают участие одновременно s- и p-электронные облака имеющие различные конфигурации. Следовательно, должны образовываться разные по прочности химические связи, т.к. при взаимодействии с другими одинаковыми атомами полнота перекрывания будет разной.
Рассмотрим образование молекулы СН4
. В возбужденном атоме углерода валентные электроны располагаются на 2s1
2p3
, т.е. по одному на каждой орбитале. (рис.4.4.).
При взаимодействии атома углерода с водородом образуется четыре ковалентных связи. Перекрывание электронных облаков водорода с р-облаками углерода происходит по полосам р-облаков, а s-облако углерода с s-облаком водорода может перекрываться в любом месте, т.к. все направления равноценны. При этом, площадь перекрывания s-облака у углерода с водородом будет отличаться от площади перекры–
 вания р-облаков. Следовательно, в молекуле СН4
один атом водорода будет иметь иную прочность связи, чем остальные три, чего практически не бывает. Все четыре атома водорода в молекуле метана неразличимы, имеют одинаковую энергию связи. Напрашивается вывод: все четыре облака в возбужденном атоме углерода имеют одинаковую форму и плотность. Эта идея привела к возникновению теории гибридизации. вания р-облаков. Следовательно, в молекуле СН4
один атом водорода будет иметь иную прочность связи, чем остальные три, чего практически не бывает. Все четыре атома водорода в молекуле метана неразличимы, имеют одинаковую энергию связи. Напрашивается вывод: все четыре облака в возбужденном атоме углерода имеют одинаковую форму и плотность. Эта идея привела к возникновению теории гибридизации.
В основе теории гибридизации лежит идея преобразо-
Рис.4.11. Расположение вания электронных облаков центрального атома перед его
валентных электронов взаимодействием с другими атомами. В результате такой
в возбужденном атоме перестройки электронные облака центрального атома раз-
углерода. ные по форме и плотности преобразуются в новые
(гибридные) облака одинаковой формы и плотности.
Так, у атома углерода в результате перестройки s-облако за счет своей плотности и частично плотности р-облаков приобретает форму односторонней гонтели. Аналогично все р-облака за счет своей плотности и остаточной плотности s-облака становятся такими же по форме и плотности.(рис.4.12.).

Pzsp-гибридизация
Py
S
Px
гибридизация ps- гибридизация
ps- гибридизация
ps- гибридизация
Рис.4.12. Перестройка (гибридизация)-электронных облаков атома углерода.
В данном преобразовании учавствуют одно s-облако и три р-облака, поэтому такая перестройка называется sp3
-гибридизацией. Как видно из рис.4.12. в результате гибридизации не только изменяется форма облаков, изменяется также взаимное расположение облаков, увеличиваются углы между новыми (гибридными) орбиталями. Гибридное состояние атома приобретает своеобразную геометрическую конфигурацию, которая и предопределяет геометрическую структуру молекулы.
 Н Н Н Н
С Н
Н Н С Н
Н
Н
Рис.3.13. Конфигурация молекулы СН4
.
После гибридизации атом углерода (рис.4.12.) получил четыре гибридных sp-облака. После взаимодействия с водородом образуется четыре одинаковых сигма sp-гибридных связи. (рис.4.13.). Молекула СН4
приобретает конфигурацию тетраэдра.
Так как гибридные облака имеют большую вытянутость в одну сторону от ядра, чем в другую, то химическая связь, образованная гибридными облаками более прочна, чем связь, образованная отдельными облаками, например, s- и p-облаками. Гибридизация связана с энергетическим выигрышем в результате образования более прочных связей и более симметричного распределения электронной плотности в молекуле.
Рассмотрим другие типы гибридизации. sp-гибридизация.
В преобразовании участвуют одно s- и одно р-облако и гибридные sp-облака становятся линейно расположенными (рис.4.14.)

S
sp-гибридизация гибридные облака
P
исходные 1800
облака
Рис.4.14. Расположение гибридных атомов при sp-гибридизации.
При sp-гибридизации молекулы имеют линейную конфигурацию. Например, молекула BeCl2
(Cl–Be–Cl).
sp2
-гибридизация.
Перестрой электронных облаков за счет одного s-облака и двух р-облаков, приводит к образованию трех sp2
-гибридных облаков, расположенных друг относительно друга под углом 1200
.

P
1200
S
Psp2
-гибридизация
исходные гибридные
облака облака
Рис.4.15. Расположение электронных оболочек при sp2
-гибридизации.
 sp2
-гибридизация дает треугольную конфигурацию молекул. Такую конфигурацию имеет, например, молекула BCl3
. Cl sp2
-гибридизация дает треугольную конфигурацию молекул. Такую конфигурацию имеет, например, молекула BCl3
. Cl
 B–Cl B–Cl
Cl
Рис.4.16. sp2
-гибриди-
зация азота и пирами-
sp3
-гибридное молекула дальная конфигура-
состояние атома азота NH3
ция молекулы NH3
.
Рассмотрим молекулы NH3
и Н2
О. В молекуле NH3
электронные облака центрального атома азота гибридизированы. Тип гибридизации sp3
. Однако во взаимодействие вступили только три гибридных орбитали, содержащих по одному электрону. На четвертой гибридной орбитали находится два электрона и поэтому она во взаимодействии с водородом не участвует. Хотя азот имеет sp3
гибридное состояние, но конфигурация молекул не тетраэдрическая, а пирамидальная, образованная как бы за счет p3
-гибридных облаков (рис.4.16.).
В молекуле Н2
О атом кислорода находится в sp3
-гибридном состоянии. Но на двух гибридных орбиталях содержится по два электрона и только две остальных, имеющих по одному электрону, вступают во взаимодействие с водородом. Получается следующая картина: при sp3
-гибридном состоянии электронных облаков атома кислорода молекула воды имеет угловую конфигурацию, образованную только за счет гибридных атомов.(рис.4.17.)

а) б) в) О
Н Н
sp3
-гибридное состояние 104,50
кислорода
Рис.4.17. sp3
-гибридное состояние кислорода (а); конфигурация молекулы Н2
О (б,в)
Зависимость пространственных конфигураций молекул от типа гибридизации дана в таблице 4.1.
Таблица 4.1.
| Тип гибридизации |
Конфигурация молекул |
Примеры |
| sp |
линейная |
BeCl2
, ZnCl2
, Co2
. |
| sp2
|
треугольная |
H2
O, H2
S. |
| sp3
|
тетраэдрическая |
BCl3
, BF3
, Co3
2–
. |
| sp3
(только p2
занята) |
угловая |
CH4
, NH4
+
, BH4
–
. |
| sp3
(только p3
занята) |
пирамидальная |
SbH3
, NH3
. |
| sp2
d |
квадратная |
PCl4
2–
. |
| sp3
d |
бипирамидальная |
PtCl5
. |
| sp3
d2
|
октаэдрическая |
SF6
. |
4.3.2. Метод молекулярных орбиталей.
К сожалению метод валентных связей, имеющий хорошую наглядность, не смог объяснить ряд особенностей отдельных молекул и устойчивость частиц. Так, метод ВС не мог объяснить, почему в молекуле O2
остаются неиспользованными два электрона и молекула обладает магнитными свойствами, почему существуют и являются достаточно устойчивыми ионы Н2
–
, Ne2
+
, O2
+
и др.? Ответ на многие “почему?” был получен после введения в теорию химической связи метода молекулярных орбиталей (метода МО).
Метод молекулярных орбиталей базируется на следующих положениях:
– Электроны в молекулах находятся на молекулярных орбиталях, как у атома – на атомных.
– молекулярные орбитали получаются при складывании атомных орбиталей.
– Из двух атомных орбиталей образуется две молекулярные орбитали, одна из которых имеет более низкую энергию.
– Орбиталь с более низкой энергией называется связывающей, а с более высокой – разрыхляющей.
– Образуются как сигма (s-), так и пи (p-) молекулярные орбитали.
– Распределение электронов по молекулярным орбиталям происходит в соответствии тех же принципов, что и по атомным: принципа наименьшей энергии, принципа Паули и правила Гунда.
       При взаимодействии двух s-атомных образуется две молекулярные орбитали: ss
св
и ss
раз
(рис.4.18.). При взаимодействии двух s-атомных образуется две молекулярные орбитали: ss
св
и ss
раз
(рис.4.18.).
 ss
раз ss
раз
+

S S ss
св
Рис.4.18. Схема образованияss
-молекулярных орбиталей.
 Р-атомные орбитали в зависимости от способа взаимодействия способны образовывать два типа молекулярных орбиталей spx
-МО и ppy
(
pz
)
-МО. (рис.4.19. и 4.20.) Р-атомные орбитали в зависимости от способа взаимодействия способны образовывать два типа молекулярных орбиталей spx
-МО и ppy
(
pz
)
-МО. (рис.4.19. и 4.20.)
spx
раз
+
PxPxspx
св
Рис.4.19. Схема образования spx
–МО.
ppz
раз
+
ppz
св
PzPz
Рис.4.20. Схема образования ppz
–МО.
Рассмотрим с позиции метода МО несколько молекул.
Молекула Н2
.
У каждого атома водорода имеется на атомных орбиталях по одному s-электрону. При взаимодействии водородов атомные орбитали объединяются и образуют, как показано на рис.4.18. две молекулярные орбитали: ss
св
и ss
раз
. Диаграмма взаимного расположения связующих и разрыхляющих молекулярных орбиталей показана на рис.4.21.
По принципу наименьшей энергии и принципу Паули оба электрона располагаются на ss
св
-орбитале. Орбиталь ss
раз
остается свободной.
 Метод МО позволяет оценивать проч- Метод МО позволяет оценивать проч-
А.О. МО А.О. ность химической связи путем расчета
Н’ Н2
H’’ кратности связи. Кратность связи (К.С.)
ss
раз
определяется как полуразность числа
электронов на связующих орбиталях (nсв
) и
числа электронов на разрыхляющих (nраз
)
1S 1S КС= nсв
– nраз
/2
ss
св
Для молекулы водорода кратность связи
Рис.4.21. Энергетическая диаграмма равна 1. КСн2
=2–0/1=1
молекулы Н2
. Энергия диссоциации молекулы Н2
состав-
ляет 432 кДж/моль.
Молекула Не2
.
Энергетическая диаграмма молекулы по методу МО представлена на рисунке 4.22.
А.О. МО А.О. По сравнению с молекулой водорода,
Не’ Не2
Hе’’ энергетическая диаграмма молекулы Не2
ss
раз
содержит также два электрона на ss
раз
-
орбите, число электронов на связующей и
разрыхляющей орбиталях одинаково.
1S 1S Кратность связи молекулы равна нулю
(КСне2
=2–2/2=0). Выигрыша энергии нет.
ss
св
Следовательно, молекула Не2
не существует.
Рис.4.21. Энергитическая схема Рассмотрим двухатомные молекулы
молекулы Не2
. элементов второго периода.
У элементов второго периода, кроме 1S-орбиталей, в образовании МО принимают участие 2S-, 2Px-, 2Py-, и 2Pz-орбитали. Комбинация 2S-атомных орбиталей дает s2
s
св
-и s2
s
раз
-орбитали. Взаимодействие 2p-орбиталей приводит к образованию двух типов МО-sр
св
-, sр
раз
- и pp
св
-, pp
раз
-орбиталей. s2
px
св
-и s2
px
раз
-молекулярные орбитали образуются от 2Px-атомных орбиталей, вытянутых вдоль оси “x”, соединяющей центры объединяющихся атомов. Так как 2Py- и 2Pz-атомные орбитали расположены перпендикулярно этой оси, следовательно они образуют ppy
св
-, ppy
раз
-, ppz
св
- и ppz
раз
-орбитали, лежащие во взаимноперпендикулярных плоскостях. Форма p-молекулярных орбиталей показана на рис.4.20.
В соответствии со спекторскопическими данными молекулярные орбитали двухатомных молекул по уровню энергии располагаются в следующий ряд:
s1
s
св
<s1
s
раз
<s2
s
св
<s2
s
раз
<s2
px
св
<p2
py
св
=p2
pz
св
<p2
py
раз
=p2
pz
раз
<s2
px
раз
.
Такой порядок расположения молекулярных орбиталей характерен для молекул второй половины периода (молекулы О2
,F2
,Nе2
).
При энергетической близости 2S- и 2P-атомных орбиталей (В, С, N) электроны на s2
s
и s2р
–орбиталях взаимно отталкиваются, поэтому p2
py
св
и p2
pz
св
–молекулярные орбитали оказываются энергетически более выгодными, чем s2
px
св
–МО. Порядок расположения молекулярных орбиталей несколько изменяется и имеет такую последовательность:
s1
s
св
<s1
s
раз
<s2
s
св
<s2
s
раз
<p2
py
св
=p2
pz
св
<s2
px
св
<p2
py
раз
=p2
pz
раз
<s2
px
раз
.
Рассмотрим более подробно несколько молекул второго периода.
Молекула
N2
.
Расположение молекулярных орбиталей представлено на рис.4.23.
 А.О. МО А.О. Электроны 1S-атомных ор- А.О. МО А.О. Электроны 1S-атомных ор-
N’ N2
N’’ биталей азота образуют s1
s
св
-
spx
раз
и s1
s
раз
-МО. Аналогично элек-
ppy
раз
ppz
раз
троны 2S-АО образуют s2
s
св
-
spx
св
и s2
s
раз
-МО. Электроны 2Р-
2P 2P подуровней азота при взаимо-
ppy
св
ppz
св
действии дают ppy
,pz
св
, spx
св
и,
соответственно, такие же раз-
рыхляющие МО. В сумме оба
2S 2S атома азота имеют 10АО, на
s1
s
раз
некоторых находится 14 элек-
тронов, молекулярных орбита-
1S 1S лей образуется тоже 10. На них
s1
s
св
должно разместиться 14 элек-
Рис.4.23. Схема расположения молекулярных тронов. Причем, заполнение
орбиталей молекулы N2
. МО электронами происходит с
соблюдением трех известных принципов. В итоге, незаполненными остались p2
py
раз
-, p2
pz
раз
- и s2
px
раз
-МО. Определим кратность связи молекулы N2
. К.С.N
2
=10-4/2=3. Молекула с кратностью связи 3 очень прочная. Энергия диссоциации этой молекулы равна 940 кДж/моль. По сравнению с молекулой Н2
(для которой К.С.=1 и энергия диссоциации равна 435 кДж/моль) молекула азота сильно повысила свою прочность.
Электронная структура молекул, аналогична электронной структуре атома, может быть изображена при помощи электронных формул. В электронных формулах указываются все МО, заполненные электронами. Например, электронная формула молекулы Н2
имеет простой вид 2Н=Н2
[(s1
s
св
)2
]. Электронная формула молекулы N2
более сложная: 2N=N2
[(s1
s
св
)2
(s1
s
раз
)2
(s2
s
св
)2
(s2
s
раз
)2
(ppy
,pz
св
)4
(spx
св
)2
].
Молекула О2
.
Атом кислорода располагается во второй половине периода, поэтому энергетическое различие между 2S- и 2Р-подуровнями больше, чем у атома азота, что не влечет ощутимое отталкивание 2S- и 2Р-электронов, поэтому последовательность в расположении МО не изменяется. Энергетическая схема орбиталей молекулы О2
показана на рис.4.24.
У атомов кислорода суммарное число орбиталей такое же, как у азота – 10, следовательно, молекулярных орбиталей у О2
– тоже десять. Суммарно количество электронов у молекулы О2
на два электрона больше, чем у молекулы азота. При распределении электронов по МО кислорода, в соответствии основным принципам распределения, последние два электрона занимают p2
py
раз
- и p2
pz
раз
-орбитали, по одному на орбиталь (правило Гунда). Наличие неспаренных электронов на МО придает молекуле кислорода новые свойства, по сравнению с молекулой азота. Молекула кислорода становится парамагнитной, т.е. она приобретает магнитные свойства и способна притягивать магнитным полем. У диамагнитных веществ все электроны парные.
Рассмотрим кратность связи в молекуле кислорода. К.С.О
2
=10-6/2=2. По сравнению с молекулой азота, молекула кислорода должна быть менее прочной. Действительно это так. Энергия диссоциации молекулы кислорода составляет 494 кДж/моль.
 АО МО AO АО МО AO
О’ O2
O’
s2
px
св
p2
py
раз
p2
pz
раз
p2
py
св
p2
pz
св
2P 2P
s2
px
св
s2
s
раз
2S 2S
s2s
св
s1s
раз
1S 1S
s1s
св
Рис.4.24. Схема расположения молекулярных орбиталей O2
.
Электронная формула молекулы кислорода:
2О=O2
[(s1
s
св
)2
(s1
s
раз
)2
(s2
s
св
)2
(s2
s
раз
)2
(s2
px
св
) (p2
py
,z
св
)4
(p2
py
,z
раз
)4
].
Рассмотрим молекулярную частицу, например молекулярный ион O+
2
. На рис.4.25. дана энергетическая схема такой частицы.
АО МО AO
О’ O+
2
O+
s2
px
раз
p2
py
раз
p2
pz
раз
p2
py
св
p2
pz
св
2P 2P
s2
px
св
s2
s
раз
2S s2s
св
2S
s1s
раз
1S s1s
св
1S
Рис.4.25. Схема расположения молекулярных орбиталей частицы O+
2
.
В молекулярном ионе кислорода, частица O+
2
, суммированный заряд ядер кислорода на единицу превышает суммированный заряд электронной оболочки молекулы. Следовательно, у частицы O+
2
на один электрон меньше, чем у молекулы кислорода O2
. На схеме молекулярных орбиталей (рис.4.25.) на pp
раз
орбиталях имеется только один неспаренный электрон (орбиталь p2
py
раз
). Это вносит некоторые изменения в свойства таких частиц по сравнению с молекулами O2
. По-видимому, ослабляют парамагнитные свойства и усиливают прочность молекулярного образования. Кратность связи O+
2
будет (К.С. O
2
+
=10-5/2=2,5) на 0,5 единиц выше. Следовательно такие кислородные частицы очень устойчивы. Энергия диссоциации O+
2
равна 629 кДж/моль.
Сравнительные данные по распределению электронов на МО, кратности связи и энергии диссоциации молекул и некоторых молекулярных частиц элементов второго периода представлены в таблице 4.2.
Таблица 4.2.
| Li2
|
Be2
|
B2
|
C2
|
N+
2
|
N2
|
O+
2
|
O2
|
O-
2
|
O2-
2
|
F2
|
Ne2
|
 spx
раз spx
раз
|
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
ppy
раз
,
ppz
раз
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
ppy
св
,
ppz
св
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
—
—
|
| spx
св
|
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
| s2s
раз
|
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
| s2s
св
|
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
| s1
s
раз
|
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
| s1
s
св
|
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
| Кратность связи |
1 |
0 |
1 |
2 |
2,5 |
3 |
2,5 |
2 |
1,5 |
1 |
1 |
0 |
| Длина связи, А |
2,67 |
— |
1,59 |
1,24 |
1,12 |
1,09 |
1,12 |
1,21 |
1,34 |
1,46 |
1,41 |
| Энергия диссоци-ации кДж/моль |
99
|
0
|
276
|
602
|
843
|
942
|
634
|
494
|
394
|
151
|
0
|
| Парамагнитные свойства |
+
|
+
|
+
|
+
|
Метод МО хорошо объясняет и молекулы и молекулярные частицы, состоящие из разных элементов.
Молекула СО.
Оба атома – углерод и кислород – имеют на внешнем уровне одинаковое количество орбиталей, но у углерода на два электрона меньше (углерод:2s2
2p2
; кислород: 2s2
2p4
).
 А.О. МО А.О. А.О. МО А.О.
C CO O
spx
раз
ppy
раз
ppz
раз
2Pspx
св
2P
ppy
св
ppz
св
s2s
раз
2S s2s
св
2S s1s
раз
1S s1s
св
1S Рис.4.26. Схема расположения молекулярных орбиталей СО.
При взаимодействии атомных орбиталей углерода с атомными орбиталями кислорода получаются молекулярные орбитали СО, аналогичные как по форме, так и по взаимному расположению, орбиталям гомоядерных молекул, например N2
. Энергетическая схема молекулы СО показана на рисунке 4.26.
В молекуле СО четырнадцать электронов. Они распределены по МО в соответствии известных трех принципов. На связующих орбиталях находится 10 электронов, а на разрыхляющих – только четыре. Кратность связи равна 3, как у молекулы N2
. Следовательно ,молекула СО должна быть очень устойчивой и, в действительности, СО напоминает молекулу N2
, энергия диссоциации СО равна 1069 кДж/моль, что на 129 кДж/моль больше, чем энергия диссоциации N2
. Можно сказать, что молекула СО изоэлектронная молекуле N2
. Электронная формула молекулы следующая: С+О=СО[(s1
s
св
)2
(s1
s
раз
)2
(s2
s
св
)2
(s2
s
раз
)2
(ppy
,z
св
)4
(spx
св
)2
].
Молекула
NO.
Энергетическая диаграмма этой молекулы схожа с диаграммой частицы O+
2
(табл.4.2.). Схема представлена на рис 4.27.
А.О. МО А.О.
N NO O
spx
раз
ppy
раз
ppz
раз
2Pspx
св
2P
ppy
св
ppz
св
s2s
раз
2S s2s
св
2S s1s
раз
1S s1s
св
1S Рис.4.27. Схема расположения молекулярных орбиталей NО.
Молекула NO имеет неспаренный электрон, она парамагнитна. Кратность связи 2,5. Электронная формула NO:
[(s1
s
св
)2
(s1
s
раз
)2
(s2
s
св
)2
(s2
s
раз
)2
(ppy
,z
св
)4
(spx
св
)2
(ppy
,z
раз
)1
].
Некоторые характеристики гетероядерных молекул и частиц, полученных из элементов второго периода даны в табл.4.3.
Таблица 4.3.
| Молекула, частица. |
Электронная конфигурация. |
Кратность связи. |
Длина
связи, А.
|
Энергия диссоциации кДж/моль |
| BN |
[C2
] |
2 |
1,28 |
385 |
| CN+
|
то же |
2 |
1,17 |
| BO |
[N2
+
] |
2,5 |
1,20 |
800 |
| CN |
то же |
2,5 |
1,17 |
756 |
| CO+
|
то же |
2,5 |
1,12 |
| NO |
[O2
+
] |
2,5 |
1,15 |
627 |
| CN-
|
[N2
] |
3 |
1,14 |
940 |
| CO |
то же |
3 |
1,28 |
1069 |
| NO+
|
то же |
3 |
1,062 |
1048 |
Многоатомные молекулы.
В качестве примера рассмотрим молекулы СН4
, NH3
, CO2
.
Молекула СН4
.
Выше было показано (4.3.1.), что атомные орбитали углерода в процессе взаимодействия с другими элементами (в частности с водородом) претерпевают перестройку, названную гибридизацией и в молекуле СН4
все связи идентичные и неразличимые. Эта особенность должна быть отражена и в методе молекулярных орбиталей. На схеме, приведенной на рисунке 4.28.,наряду с АО углерода показаны гибридные орбитали.
Е А.О. А.О. МО А.О.
С исходн. С гибр. СН4
4sх
раз
4Н
2Р
4sх
св
(или sсв
s
,
x
,
y
,
z
)
2SP3
1S
1S ss
*
Рис.4.28. Схема расположения молекулярных орбиталей СН4.
1S-атомная орбиталь углерода, содержащая два электрона как орбиталь, лежащая в глубине атома, участие в образовании связи не принимает. Она без изменения переходит в молекулярную несвязывающую орбиталь. На рис.4.28. она обозначена ss
*
. В образовании МО принимает участие 8 атомных орбиталей (четыре от атома углерода и четыре от четырех атомов водорода). Следовательно, образуется 8 молекулярных орбиталей: 4 – связывающих и 4 – разрыхляющих. Заполняются электронами только все связывающие орбитали. Так как направление всех связей между атомом углерода и каждым из водородов совпадают с линией, связывающей центры атомов, и по прочности одинаковы, то все связывающие молекулярные орбитали находятся на одном и том же энергетическом уровне и обозначены одинаково sх
св
. Аналогично и разрыхляющие. Электронная формула молекулы может быть записана так: СН4
[4*(sх
св
)2
].
Молекула
NH3
.
 В отличие от СН4
в молекуле NH3
имеется еще одна несвязывающая орбиталь s2
s
*
, т.к. 2s-атомная орбиталь азота практически не принимает участия в образовании связей. Схема расположения МО NH3
представлена на рис. 4.29. В отличие от СН4
в молекуле NH3
имеется еще одна несвязывающая орбиталь s2
s
*
, т.к. 2s-атомная орбиталь азота практически не принимает участия в образовании связей. Схема расположения МО NH3
представлена на рис. 4.29.
Е АО МО АО
NNH3
3H
sх,
y
,
z
раз
2Psх,
y
,
z
св
(или 3sх
св
) 2P
2S s2s
*
1S s1s
*
Рис.4.29. Схема расположения МО в молекулеNH3
.
Электронная конфигурация молекулы NH3
:N+3H=NH3
[(ss
св
)2
(sy
)2
(sz
)2
] или [3(sx
)2
].
Кратность связи в таких многоатомных молекулах определяется по известному принципу, но затем необходимо разделить на число связей. К.С. N-H
=(6-0/2):3=1
Молекула СО2
.
 При взаимодействии углерода с кислородом атомные орбитали углерода претерпевают гибридизацию. На рисунке 4.30. показана схема расположения МО в молекуле СО2
. На этой схеме не указаны 1s атомные орбитали углерода и обеих кислородов, т.к. они являются внутренними, во взаимодействие не вступают, остаются несвязывающими орбиталями. При взаимодействии углерода с кислородом атомные орбитали углерода претерпевают гибридизацию. На рисунке 4.30. показана схема расположения МО в молекуле СО2
. На этой схеме не указаны 1s атомные орбитали углерода и обеих кислородов, т.к. они являются внутренними, во взаимодействие не вступают, остаются несвязывающими орбиталями.
АО АО МО АО
С исходн. С*гибрид. СО2
2О
sх
раз
py
,
z
раз
2Pyz
2P 2Px,y,z
p*y,z
2S 2SPx
sх
св
py
со
,z
2S
ss
*
Рис.4.30. Схема расположения МО в молекуле СО2
.
Несвязующими становятся еще четыре орбитали. Они переходят в молекулу СО2
от двух атомов кислорода (две от 2s-атомных орбиталей и две от 2p-АО). Электронная структура молекулы может быть представлена следующей электронной формулой:
C+2O=СО2
[(ppy
,z
св
)4
(ss
,
x
св
) 4
(p*py
,
z
)4
]. K.C.c-o
=(8-0/2):2=2
 Ион СО2
2-
.
Схема молекулярных орбиталей в карбонат-ионе СО2
2-
показана на рис.4.31. Ион СО2
2-
.
Схема молекулярных орбиталей в карбонат-ионе СО2
2-
показана на рис.4.31.
Е АО АО МО АО
С исход. С*гибрид. СО2
2-
sх
раз
3О
pz
раз
2Px,y,z 2Pz
py
*
2S 2Px,y,z
sх
св
pz
св
ss
* 2S
Рис.4.31. Схема расположения МО в карбонат-ионе СО2
2-
.
В карбонат-ионе несвязующими МО являются орбитали полученные от 2S-АО трех атомов кислорода (их три: ss
*), а также три орбитали ppy
*, полученные в результате превращения трех 2Р-атомных орбиталей кислорода. На МО карбонат-иона находятся два неспаренных электрона, расположенных на pz
-разрыхляющих орбиталях. Электронная формула карбонат-иона следующая:
C+3O+2e=СО2
2-
[(pz
св
)2
(pz
св
)2
(sх
св
)2
(sх
св
)2
(sх
св
)2
(pz
x
)6
(pz
,
z
раз
)2
].
Кратность связи составляет (10-2/2):3=1,33.
Подобную структуру имеет нитрат-ион NО3
-
. Он является изоэлектроном иону СО2
2-
.
Из рассмотренного можно сделать следующий вывод относительно основных характеристик ковалентной связи.
Ковалентная связь.
1). Обладает полярностью.
2). Имеет направленность.
3). Стремится к насыщаемости.
4). Является очень прочной.
Молекулы химических соединений, образованные ковалентной связью, имеют определенный количественный и качественный состав (например, молекула воды состоит из одного атома кислорода и двух атомов водорода. 88,9% от массы молекулы воды приходится на кислород и 11,1% - на водород). Ковалентные молекулы характеризуются конкретной геометрической конфигурацией (молекулаСН4
-тетраэдр, молекула BeCl4
-линейная). Ковалентные молекулы могут проявлять либо парамагнитные, либо диамагнитные свойства. Химические соединения с ковалентной связью могут иметь разные агрегатные состояния: твердое, жидкое и газообразное (CuO-твердый, TiCl4
-жидкость, SO2
-газ).
4.4 Ионная связь.
Само название связи – ионная – указывает на то, что связь возникает в результате взаимодействия ионов.
Ионной называют такую связь, которая образуется между катионом и анионом в результате их электростатического взаимодействия.
Правомерно встает вопрос, когда химические элементы могут превращаться в положительно и отрицательно заряженные ионы (катионы и анионы).
Рассматривая полярную ковалентную связь выяснили, что область перекрывания орбиталей взаимодействующих атомов (т.к. область перекрывания орбиталей дает увеличение электронной плотности создаваемой парой электронов, для простоты будем использовать понятие “электронная пара”), общая электронная пара смещается в сторону более электроотрицательного элемента. Величина полярности молекулы оценивается дипольным моментом m. Из расчета дипольного момента полярных молекул выяснили, что при m >4,8Д полярная ковалентная связь превращается в ионную. В этом случае электрон внешней оболочки одного атома переходит на оболочку более электроотрицательного атома. Такой переход возможен при взаимодействии атома лития с атомом фтора. Это показано на рис.4.32.
Под действием поля атома фтора один электрон из внешней оболочки лития переходит на оболочку фтора. Оба атома приобретают устойчивые оболочки (литий – двухэлектронную оболочку, фтор - восьмиэлектронную оболочку), но при этом оба атома становятся ионами: литий – положительно заряженным – катионом, а фтор - отрицательно заряженным – анионом. Относительная электроотрицательность лития равна 1 (Е
Li
=1), а фтора – 4 (Е
F
=4). Разность в электроотрицательностях составляет три единицы(DЕ
=3). Считается, что полярная ковалентная связь превращается в ионную при разности в электроотрицательностях взаимодействующих атомов более 1,9 единицы.

+3 + +9 +3 + +9
Li F Li+
F—
+ + —+ —
Li+
F—
LiF
Рис.4.32. Образование ионной молекулы LiF.
 Следовательно, эта величина как бы является критерием образования ионной связи. DЕ
>1,9. Следовательно, эта величина как бы является критерием образования ионной связи. DЕ
>1,9.
Более точной оценкой степени ионности полярной связи может быть расчет отношения заряда диполя q к заряду электрона е: q/e. Cделаем оценку степени ионности связи в молекуле HCl. При длинные связи l=1,27 А0
и величине заряда диполя q=e=4,8*1010
эл.ст.ед. Расчетное значение дипольного момента будет: mтеор.
=l*q= =1,27*10-8
* 4,8*10-10
=6,11Д. Экспериментально определенное значение mэкс.
=1,039 Д.
значение mэкс.
=1,039 Д. Тогда отношение q к е составит: q/e= mэкс.
/ mтеор.
*100=17%
Степень ионности связи в молекуле НCl всего 17%. Остальные 83% составляет ковалентный характер связи. Обычно считают, что связь является приемущественно ионной, когда степень ионности q/е>50%. При критерии DЕ
>1,9 молекула приобретает степень ионности более 50%.
100%-ной ионной связи практически не бывает. Для 100%-ной ионной связи необходимо, чтобы разность в электроотрицательностях элементов DЕ
достигла величины 3,5. На самом деле, наиболее крайние по электроотрицательности элементы франция (Fr) и фтора (F) имеют всего DЕ
=3,3.
 На рис.4.33. дана зависимость степени ионности связи (q/e) от разности в электроотрицательностях атомов DЕ
. На рис.4.33. дана зависимость степени ионности связи (q/e) от разности в электроотрицательностях атомов DЕ
.
q/e*100% 100
Рис.4.33. зависи- 90 ионные связи
мость степени 80
ионности связи 70
от разности 60
электроотрица- 50
тельности атомов. 40 ковалентные
30 связи
20
10
0,4 0,8 1,2 1,6 2,0 2,4 2,8 3,2 3,6DЕ
Рассмотрим свойства ионной связи на примере хлорида натрия. В следствии противоположности зарядов оба иона Na+
и Сl-
притягиваются друг к другу. Однако, сблизившись до определенного предела, они останавливаются на оптимальном расстоянии (r0
), при котором притяжение уравновешивается взаимным отталкиванием их электронных оболочек.
Положительно и отрицательно заряженные ионы, образующие ионное соединение, представим в виде заряженных шариков, силовые поля которых равномерно распределяются в пространстве во всех направлениях (рис.4.34.)
 Рисунок 4.34. наглядно показы- Рисунок 4.34. наглядно показы-
вает, что ионы держатся друг возле
+ —друга за счет силового электростати-
ческого поля. Насыщаемость силовых
линий происходит только в области
Рис.4.34. Распределение электрических между ионами, а по бокам каждого ио- силовых полей двух разноименных ионов. на силовые линии не компенсируются.
Отсюда первое свойство ионной связи – связь ненасыщенная.
Ненасыщаемость ионной связи приводит к тому, что все ионные соединения кристаллические вещества с высокими температурами плавления и кипения. Только в кристаллическом соединении за счет образования определенной кристаллической решетки, в которой каждый ион окружен рядом ионов противоположного знака, происходит компенсация силовых линий. Кристаллическая решетка NaCl построена из двух гранецентрированных кубических подрешеток, состоящих одна из ионов Na+
, другая из ионов Сl-
, сдвинутых одна в другую на половину ребра куба. Число, показывающее, сколько ионов противоположного знака окружает данный ион в кристалле, называется координационным числом. Координационное число зависит от размеров ионов и определяется соотношением радиусов ионов. Так, при соотношении радиусов ионов в пределах 0,41-0,73 оно равно 6, а при соотношении радиусов ионов 0,73-1,37 координационное число равно 8. Координационное число решетки NaCl равно 6, это значит, что ион натрия (в кристалле NaCl) окружает шесть ионов хлора и, наоборот ион хлора окружает шесть ионов натрия.
Когда мы изображаем хлорид натрия ( поваренную соль) символом NaCl, мы допускаем определенную неточность. Следовало бы писать NaCl-кристалл или (NaCl)n
. Даже в парообразном состоянии хлорид натрия наряду с одинарными молекулами присутствуют ассоциаты (NaCl)2
и (NaCl)3
.
Второе свойство ионной связи заключается в том, что она ненаправлена. Нельзя указать направление, по которому ион хлора подходит к иону натрия, у каждого иона все направления равноценны, и с любой стороны один ион может подойти к другому. Если сравнить с ковалентной связью, в ковалентных молекулах взаимодействие между атомами происходит в направлении распространения электронного облака и ковалентные молекулы имеют определенную геометрическую конфигурацию, ионные молекулы такого свойства не имеют.
Как было сказано, вещества, образованные ионными молекулами, при обычных условиях являются твердыми кристаллическими веществами с высокими температурами плавления. (tпл
NaCl – 800 0
C; tпл
NaF – 995 0
C). Это свидетельствует о прочности связи в кристалле. Энергия ионной связи (Есв(ион.)
) велика. Кроме энергии электростатического взаимодействия Еэ
, энергия связи (Есв
) включает в себя энергию сродства к электрону (Еср
) неметалла и энергию ионизации атома металла (I). Расчет показывает, что энергия ионной связи молекулы NaCl равна 422,6 кДж/моль.
ЕNaCl
= Еэ
- Еср
+ I =5,7-5,1+3,7= 4,3 эВ=422,6 кДж/моль.
Но прежде, чем разорвать связь между атомами в молекуле NaCl, необходимо разрушить кристаллическую решетку этого вещества. Следовательно, требуется дополнительная затрата энергии. Расчеты показывают, энергия кристаллической решетки (NaCl)n
равна 764 кДж/моль.
Ионная связь возникает не только между простыми ионами, она реализуется и между сложными ионами: катионами NH4
+
, [Cu(NH3
)4
]+
и анионами NO3
-
, SO4
2-
, CO3
2-
, [PtCl6
]2-
т.д.
За единицу валентности атомов в ионных соединениях принимают единичный заряд иона. Например, в ионной молекуле NaCl атом натрия имеет заряд (+1), а атом хлора (-1). Следовательно, оба атома одновалентны.
4.5. Донорно – акцепторная связь.
 Рассматривая ковалентную связь, выяснили, что она образуется в результате перекрывания одноэлектронных валентных облаков взаимодействующих атомов. Так, атом азота взаимодействует тремя одноэлектронными облаками, например, с тремя атомами водорода, образуя молекулу аммиака NH3
. H Рассматривая ковалентную связь, выяснили, что она образуется в результате перекрывания одноэлектронных валентных облаков взаимодействующих атомов. Так, атом азота взаимодействует тремя одноэлектронными облаками, например, с тремя атомами водорода, образуя молекулу аммиака NH3
. H
H:N:
H
Но атом азота имеет на внешнем квантовом уровне еще два электрона, расположенных на 2s-подуровне, и для их распаривания у атома возможности нет. Возникает вопрос: может ли пара электронов, находящихся на 2s-орбитале, образовывать химическую связь? Оказывается, что может. Но для этого нужны определенные условия, а именно, наличие свободной орбитали.
Такое взаимодействие, т.е. взаимодействие за счет пары электронов (ее называют “неподеленной”) одного атома и свободной орбитали другого атома называют донорно-акцепторным, а химическую связь, образованную при помощи этого взаимодействия – донорно – акцепторной связью.
Следовательно, необходимым условием для образования донорно – акцепторной связи, является наличие неподеленной электронной пары одного атома (молекулы) и свободной орбитали другого атома (молекулы). Неподеленная пара донора занимает свободную орбиталь акцептора. Образуется двухэлектронная орбиталь, которая обслуживает оба атома (атом донора и атом акцептора).
Если обратиться к аммиаку, то молекула NH3
может образовывать донорно – акцепторную связью с любой другой молекулой, у которой атомы имеют свободные орбитали.
 Например, NH3
легко взаимодействует с молекулой HCl. В полярной молекуле HCl общая электронная пара (область перекрывания орбиталей) сильно смещена в сторону более электроотрицательного атома хлора. При этом орбиталь атома водорода практически свободна и она может принимать электронную пару донора (атома азота) образуя дополнительную связь. H H Например, NH3
легко взаимодействует с молекулой HCl. В полярной молекуле HCl общая электронная пара (область перекрывания орбиталей) сильно смещена в сторону более электроотрицательного атома хлора. При этом орбиталь атома водорода практически свободна и она может принимать электронную пару донора (атома азота) образуя дополнительную связь. H H
H – N: + H:Cl H – N : H Cl
HH
В результате донорно – акцепторного взаимодействия двух нейтральных молекул NH3
и HCl получается новое соединение – хлорид аммония.
NH3
+HCl = NH4
Cl.
 Хлорид аммония – ионное соединение. Донорно – акцепторное взаимодействие превратило молекулу аммиака в ион аммония NH4
+
. В ионе аммония три водорода связаны с азотом по ковалентному принципу, а четвертый водород – по донорно – акцепторному. H + Хлорид аммония – ионное соединение. Донорно – акцепторное взаимодействие превратило молекулу аммиака в ион аммония NH4
+
. В ионе аммония три водорода связаны с азотом по ковалентному принципу, а четвертый водород – по донорно – акцепторному. H +
H – N– – –H
H донорно – акцепторная связь.
Донорно – акцепторную связь обычно обозначают пунктирной линией (– – –).
По донорно – акцепторному принципу легко взаимодействуют такие две нейтральные молекулы: BF3
и HF. У бора есть свободная орбиталь на 2р-подуровне, а у фтора неподеленная пара электронов. Бор является акцептором, а фтор донором.
F F –
F – B + :F - H F – B : F H+
. Образуется сложный (комплексный)
FF анион [BF4
]–
.
Новая более сложная молекула Н[BF4
] вобрала в себя три вида связей: между комплексным анионом BF4
–
и катионом H+
– ионная связь. В анионе BF4
–
три фтора соединены с бором по ковалентному принципу, а четвертый фтор – связан с бором посредством донорно – акцепторного взаимодействия. F –
F – B – – – F
F
Как видно из рассмотренных примеров донорно – акцепторная связь объединяет простые молекулы в комплексы. Например, ZnSO4
легко взаимодействует с аммиаком с образованием комплексного соединения – сульфата тетраамминцинка.
ZnSO4
+ 4NH3
= [Zn(NH3
) 4
]SO4
H3
N NH3
2+
Zn2+
+ 4NH3
= Zn
H3
NNH3
Наряду с межмолекулярной, донорно – акцепторная связь иногда встречается как дополнительная связь внутри одной молекулы (внутримолекулярная)
Рассмотрим два примера:
Молекула СО.
 В невозбужденном состоянии атом углерода имеет два неспаренных электрона на р–подуровне и свободную р–орбиталь. Атом кислорода на р–подуровне содержит два неспаренных электрона и одну электронную пару. В невозбужденном состоянии атом углерода имеет два неспаренных электрона на р–подуровне и свободную р–орбиталь. Атом кислорода на р–подуровне содержит два неспаренных электрона и одну электронную пару.
С + O O
s
pC
Неспаренные электроны углерода и кислорода образуют в молекуле СО две дополнительных s– и p–связи и одну донорно – акцепторную – за счет неспаренной электронной пары кислорода (донор) и свободной орбитали углерода (акцептор).
Образование дополнительной донорно – акцепторной связи в СО приводит к тому, что молекула СО очень прочная. Энергия диссоциации СО, как было указано раньше, составляет 1069 кДж/моль.
Молекула С
l2
.
Атом хлора в невозбужденном состоянии имеет на внешнем уровне один неспаренный электрон и полностью свободный d–подуровень.
spd
    При взаимодействии двух атомов хлора друг с другом их неспаренные электроны образуют s–ковалентную связь, и наряду с ней каждый атом отдает свою электронную пару на свободную орбиталь другого атома, образуя две дополнительных донорно – акцепторных связи. Следовательно, в молекуле Сl2
не одинарная, а тройная связь. Cl––Cl. Донорно – акцепторные связи При взаимодействии двух атомов хлора друг с другом их неспаренные электроны образуют s–ковалентную связь, и наряду с ней каждый атом отдает свою электронную пару на свободную орбиталь другого атома, образуя две дополнительных донорно – акцепторных связи. Следовательно, в молекуле Сl2
не одинарная, а тройная связь. Cl––Cl. Донорно – акцепторные связи
 Ковалентная связь Ковалентная связь
Энергия донорно – акцепторной связи колеблется в широких пределах: в среднем от 10 кДж/моль до 200 кДж/моль. Например, энергия диссоциации J2
–CH3
OH равна 8 кДж/моль, а энергия диссоциации AlCl3
–NH3
равна 233 кДж/моль.
Для ряда прочных комплексных соединений энергия донорно – акцепторной связи по величине близка к энергии ковалентной связи.
Так как донорно – акцепторная связь по механизму взаимодействия и по прочности близка к ковалентной связи, ее иногда причисляют к частному случая ковалентной связи.
4.6. Водородная связь.
Как и донорно – акцепторная связь, водородная связь относится к дополнительным типам связи и приводит к образованию молекулярных группировок из одинаковых или разных молекул (межмолекулярных) или связывает между собой отдельные части, функциональные группы (атомы) одной молекулы (внутремолекулярная).
Образование водородной связи обусловлено спецификой водорода как элемента, состоящего из протона и одного единственного электрона. В тех случаях, когда водород соединен с более электроотрицательным элементом, его единственный электрон смещается к более электроотрицательному элементу. Водород, лишенный электрона, превращается в особого рода положительно заряженную частицу с уникальными свойствами. Эта частица, не имеющая электронов, в отличие от остальных катионов не отталкивается электронными оболочками других частиц, а испытывает только притяжение, кроме того, атом водорода, лишенный электрона (протон), имеет ничтожно малые размеры, он может глубоко внедряться в электронную оболочку отрицательного плояризованного атома. Все это приводит к тому, что атом водорода способен образовывать дополнительную связь, которая получила название водородная. Чем больше электрон водорода оттянут в сторону атома, связанного с ним ковалентно, тем сильнее протон притягивается к электронной оболочке другого атома.
 Например, водородная связь возникает между молекулами фтористого водорода. Во вториде водорода общая электронная пара сильно смешена к атому фтора H : F, водород, почти полностью лишившись электронной плотности, притягивается к атому фтора другой молекулы фтористого водорода образуя с ним водородную связь. Эту связь обозначают тремя точками H – F…
H – F…
H – F. Например, водородная связь возникает между молекулами фтористого водорода. Во вториде водорода общая электронная пара сильно смешена к атому фтора H : F, водород, почти полностью лишившись электронной плотности, притягивается к атому фтора другой молекулы фтористого водорода образуя с ним водородную связь. Эту связь обозначают тремя точками H – F…
H – F…
H – F.
ковалентная связь водородная связь
Водородная связь по прочности выше ван–дер–*** сил, но значительно слабее в 10–15 раз ковалентной связи.
Водородная связь проявляется тем сильнее, чем больше электроотрицательность атома–партнера и чем меньше его размеры. Поэтому она наиболее характерна прежде всего для соединений фтора и кислорода, в меньшей степени для соединений азота, хлора, серы.
Энергия водородной связи зависит как от вида и состояния атома–партнера, так и от того, с какими атомами последний соседствует. Так, энергия водородной связи H…
N равна 8 кДж/моль; связь H…
О – около 21 кДж/моль, а связь H…
F приблизительно 36 кДж/моль.
Водородная связь возникает как между молекулами – межмолекулярная (H – F…
H – F; H – F…
H2
О) так и внутри отдельных молекул – внутремолекулярная.
 Н Н
С
О
ОН
Межмолекулярная водородная связь может образовываться как между молекулами одного и того же вещества (H – F…
H – F; H2
О…
H2
О; NH3
…
NH3
), так и между молекулами различных веществ (H3
N…
H2
О; HF…
H2
О).
Водородная связь между молекулами аммиака и воды приводит к образованию гидрата аммиака H3
N.
H2
О. В случае молекул аммиака и хлористого водорода межмолекулярное взаимодействие сопровождается ионизацией HCl и переходом водородной связи в донорно – акцепторную.
Ассоциация молекул, обусловленная образованием водородной связи, происходит в газообразном состоянии веществ, жидкостях и твердой фазе. Так, в парах фтористого водорода существуют молекулы (HF)n, где n=4,5 и даже 6. Этот полимер имеет следующую структуру.
 F F F F
H H H H H
FFF
Образованием водородной связи можно объяснить существование очень устойчивого аниона НF2
–
, состоящего из двух ионов F–
, соединенных протоном (F…
H – F)–
. Аналогичный ион НCl2
–
мало cтабилен, так как электроотрицательность хлора Е
С
l
=3 значительно ниже электроотрицательности фтора Е
F
=4.
 Благодаря водородной связи, например, муравьиная кислота в газообразной фазе существует в виде димера. O…
H–O Благодаря водородной связи, например, муравьиная кислота в газообразной фазе существует в виде димера. O…
H–O
H–C C–H
O–H…
O
В жидкой фазе ряд органических кислот (муравьиная, уксусная, хлоруксусная) образуют ассоциаты двух типов: цепи и циклические.
В твердом состоянии все эти кислоты образуют цепные молекулы.
…
O
C–CH2
Cl
O…
HO
ClH2
C–C
OH…
O
C–CH2
Cl
O…
HO
ClH2
C–C–OH…
Cпособность молекул к ассоциации отличает воду, аммиак, спирт и другие жидкости от неассоциированных жидкостей, например, углеводородов. Ассоциация приводит к повышению температуры плавления, температуры кипения, теплоты парообразования, изменению растворяющей способности и т.д. Если бы вода не образовывала ассоциатов, то имела бы температуру замерзания –100 0
С, а температуру кипения –80 0
С. (рис.4.35.)
Водородные связи в структуре воды и льда играют важную роль. В кристалле льда (Н2
О)n каждая молекула воды тетраэдрически соединена водородными связями с четырьмя ближайшими к ней молекулами. Такую структуру в плоскостном изображении можно представить следующей схемой.
Н

Н–О
Н Н
О–Н…
О–Н…
О–Н
Н Н–О
Н
Это создает ажурную структуру, далекую от плотной упаковки. Поэтому лед имеет небольшую плотность и значительную рыхлость.
 При плавлении льда водородные связи частично разрушаются (примерно на 10%). Это несколько сближает молекулы, поэтому вода плотнее льда. Нагревание воды, с одной стороны, приводит к ее расширению, т.е. к увеличению объема, с другой стороны, вызывает дальнейшее разрушение водородных связей и тем самым уменьшает объем. В результате плотность воды проходит через максимум при температуре +4 0
С. Считают, что при +4 0
С вода содержит ассоциаты из двух молекул (Н2
О)2
, связанных двумя водородными связями. О…
Н Н При плавлении льда водородные связи частично разрушаются (примерно на 10%). Это несколько сближает молекулы, поэтому вода плотнее льда. Нагревание воды, с одной стороны, приводит к ее расширению, т.е. к увеличению объема, с другой стороны, вызывает дальнейшее разрушение водородных связей и тем самым уменьшает объем. В результате плотность воды проходит через максимум при температуре +4 0
С. Считают, что при +4 0
С вода содержит ассоциаты из двух молекул (Н2
О)2
, связанных двумя водородными связями. О…
Н Н
Н Н…
О
Которые и обуславливают наибольшую плотность воды (равную 1г/см3
) при +4 0
С. Дальнейшее повышение температуры приводит к расширению воды и к разрыву водородной связи. Молекулы водяного пара почти не ассоциированны.
Водородная связь играет большую роль в процессах растворения. Растворимость веществ во многом зависит от их способности образовывать водородные связи с растворителем.
Водородная связь проявляется почти повсеместно: и в неорганических веществах, и в органических, в белках, в полимерах, в живых организмах. Предполагают, что и действие памяти связано с хранением информации в конфигурациях с Н–связями. Поэтому в последнее время водородная связь стала объектом дополнительного исследования.
4.7. Металлическая связь.
Металлическая связь по своей модели существенно отличается от ионной и ковалентной: она характеризуется взаимодействием положительных ионов кристаллической решетки металла и свободных электронов, не связанных с определенными ионами, свободно перемещающихся в пределах кристаллической решетки. Положительно заряженные ионы металлов располагаются в узлах кристаллической решетки, а между ними находятся как бы “коридоры”, заполненные свободными электронами, перемещающимися по всему объему металла (рис.3.36.).
Металлическая связь возможна и в жидком, и в твердом состоянии веществ.

Рис.3.36. Схема металлической решетки.
Экспериментальные изучения металлических решеток показывают, что металлы имеют простую, но плотную структуру, каждая частица окружена шестью или восемью другими. Так, в натрии единственный валентный электрон должен принимать участие в восьми связях, т.е. электрон не локализован. Каждый электрон перемещается в кристалле, и каждая частица окружена электронами, которые не принадлежат исключительно и постоянно ей. Металлическую решетку натрия можно представить в виде решетки из ионов Na+
, которая погружена в облако свободных электронов. Связь обеспечивается электростатическим притяжением между положительными ионами и электронным облаком. Такая модель позволяет объяснить некоторые свойства металла. Металлическая связь слабее ковалентной связи; металлическую решетку можно деформировать (тягучесть, ковкость металлов), но вырвать атом из такой решетки трудно; об этом свидетельствуют, в частности, высокие температуры кипения металлов: 3570
С (Hg), 8800
С (Na) и 30000
С (Fe) и т.д.
Электронное облако внутри решетки легко приводится в движение с помощью электрического поля; следовательно, металл – хороший проводник электричества, однако вырвать электрон из металла трудно, поскольку между группировкой положительных ионов и электронами действуют значительные силы притяжения.
Большая тепловая проводимость металлов тоже объясняется облаком подвижных электронов. Если часть металла нагрета, то кинетическая энергия электронов в этой области возрастает. Электроны распространяются по всему металлу, таким образом, растет температура во всей решетке.
Рассмотрим металлическую связь с позиций метода молекулярных орбиталей.
Атомы металлов содержат на внешних квантовых уровнях мало электронов и много вакантных орбиталей. При объединении атомов в кристаллическую решетку атомные орбитали металлов объединяются в молекулярные. Число молекулярных орбиталей равняется сумме орбиталей отдельных атомов. Если бы учитывались только s- и р-орбитали внешних уровней, то в этом частном случае при содержании в кристалле числа атомов (только в 1см3
N=1022
– 1023
атомов) образуется 4N молекулярных орбитали. В каждом атоме одна s-орбиталь и три р-орбитали, следовательно общее число орбиталей (1+3)N=4N.
При учете d- и f-атомных орбиталей число молекулярных орбиталей увеличивается на (5N+7N), т.е. резко возрастает.
Валентные электроны заполняют молекулярные орбитали металлов в порядке возрастания энергии. Так как молекулярных орбиталей больше, чем валентных электронов, то только часть молекулярных орбиталей занята элктронами, ее называют валентной зоной. Совокупность уровней, расположенных выше валентной зоны, и содержащая валентные орбитали, названа зоной проводимости. Благодаря близости расположения зон (валентной и проводимости) электроны легко переходят с орбиталей валентной зоны на орбитали зоны проводимости осуществляя при этом между атомами металла нелокализованные связи.
4.8. Слабые межмолекулярные взаимодействия.
 Наряду с межмолекулярными водородной и донорно – акцепторной связями между молекулами отдельных соединений могут возникать слабые межмолекулярные взаимодействия. Межмолекулярное взаимодействие приводит к тому, что газообразные вещества при соответствующих условиях могут переходить в жидкое и твердое агрегатное состояние. Наряду с межмолекулярными водородной и донорно – акцепторной связями между молекулами отдельных соединений могут возникать слабые межмолекулярные взаимодействия. Межмолекулярное взаимодействие приводит к тому, что газообразные вещества при соответствующих условиях могут переходить в жидкое и твердое агрегатное состояние.
dD
0
Рис.3.37. Результирующая кривая
межмолекулярного взаимодействия.
Силы притяжения или сцепления между молекулами называют ван–дер–Вальсовыми силами, по имени голландского ученого Ван–дер–Вальса, изучавшего межмолекулярное взаимодействие.
Межмолекулярное взаимодействие зависит прежде всего от расстояния между центрами взаимодействующих молекул. На больших расстояниях ван–дер–вальсовы силы ничтожно малы и начинают проявляться лишь на расстояниях порядка 10А. Если две молекулы приближать друг к другу, то на определенном расстоянии между ними начинают действовать силы притяжения и отталкивания. Соотношение между этими двумя силами можно выразить результирующей кривой (рис.3.37.). Силы притяжения между двумя сближающимися молекулами сначала растут, достигают некоторого максимума, а затем резко уменьшаются в следствии сильного возрастания сил отталкивания. Расстояние между молекулами d0
отвечает равновесному состоянию, когда силы притяжения и отталкивания двух сближающихся молекул уравновешиваются, d0
при этом равно 4–7А. Энергия межмолекулярного взаимодействия невелика и составляет около 8–47 кДж/моль, т.е. в 10–100 раз меньше энергии обычного химического взаимодействия.
Молекулы как валентнонасыщенные частицы не могут образовывать между собой валентных связей. Какова тогда природа ван–дер–Вальсовых сил?
 В А В А
ldl
ldl
Рис.3.38. Ориентационное дипольное
взаимодействие молекул.
ll
Так как молекулы можно разделить на полярные и неполярные, то возможны три типа взаимодействий:
а) между полярными молекулами (дипольное);
б) между полярной и неполярной молекулами (индукционное);
в) между неполярными молекулами (дисперсионное).
Рассмотрим каждый из этих типов взаимодействий.
Диполное взаимодействие (ориентационное)
– это взаимодействие двух полярных молекул. Сущность его сводится к тому, что положительный коней одной молекулы А притягивает к себе отрицательный конец другой В. Переориентировка диполей протекает до тех пор, пока притяжение между ними не уравновесится силами отталкивания (рис.3.38.). В результате взаимодействия диполей потенциальная энергия системы уменьшается, это равносильно усилению связи между молекулами. Чем больше длина диполей l взаимодействующих молекул, тем больше энергия дипольного взаимодействия. Так как тепловое движение молекул нарушает ориентацию, то естественно повышение температуры ослабляет связи ориентационного (дипольного) взаимодействия.
  Индукционное взаимодействие
– взаимодействие полярной и неполярной молекул. В неполярной молекуле значение постоянного дипольного момента равно нулюmр
=0. Под действием электрического поля полярной молекулы может индуцироваться в неполярной молекуле диполь с mi
=0 и и последняя становится индуцированно–полярной. Между постоянным диполем молекулы А индуцированным диполем молекулы В возникает индуционное взаимодействие (рис.42.). Не все полярные молекулы обладают одинаковой способностью к индуцированию: чем выше поляризуемость молекулы, тем больше величина возникающего в ней индуцированного момента и тем сильнее индукционное взаимодействие. Индукционное взаимодействие
– взаимодействие полярной и неполярной молекул. В неполярной молекуле значение постоянного дипольного момента равно нулюmр
=0. Под действием электрического поля полярной молекулы может индуцироваться в неполярной молекуле диполь с mi
=0 и и последняя становится индуцированно–полярной. Между постоянным диполем молекулы А индуцированным диполем молекулы В возникает индуционное взаимодействие (рис.42.). Не все полярные молекулы обладают одинаковой способностью к индуцированию: чем выше поляризуемость молекулы, тем больше величина возникающего в ней индуцированного момента и тем сильнее индукционное взаимодействие.
А В
ll=0 l=0 l=0
l li
li
li
Рис.42. Индукционное взаимо– Рис.43. Дисперсионное взаимо–
действие молекул. действие молекул.
Так как индуцирование приводит к изменению или деформации электронной оболочки молекулы, то этот тип взаимодействия иногда называют деформационным.
Индуцирование или деформация неполярной молекулы зависит от напряженности поля полярной молекулы, а поэтому индуцированный эффект не зависит от температуры.
Дисперсионное взаимодействие
– взаимодействие двух неполярных молекул. Хотя у обеих неполярных молекул дипольный момент равен нулю, вследствии пульсирующего движения электронного облака (или движения электронов внутри молекулы) в одной из молекул на мгновение возникает незначительный дипольный момент, который индуцирующе действует на соседнюю молекулу, и т.д. Между этими диполями возникает дисперсионное взаимодействие (рис.43.), которое тем больше, чем легче поляризуется молекула или атом и чем меньше расстояние между взаимодействующими молекулами. На дисперсионном взаимодействии основан процесс сжижения благородных и двухатомных элементарных газов, молекулы которых не имеют дипольного момента.
Следует отметить, что для реальных молекул установить какой-либо единственный тип взаимодействия невозможно. Практически при взаимодействии молекул проявляются в определенной степени все три типа взаимодействия. Вклад каждого из рассмотренных типов межмолекулярного взаимодействия зависит в основном от двух свойств взаимодействующих молекул: полярности и поляризуемости (деформируемости). Чем выше полярность, тем значительнее роль ориентационных сил; чем больше деформируемость, тем значительнее роль дисперсионных сил. Индукционные силы зависят от обоих факторов.
Все три типа сил межмолекулярного взаимодействия имеют одну и ту же природу – электростатическую и обуславливаются электрическими полями молекул или атомов.
Глава 5.
Агрегатные состояния химических веществ.
В химии, а еще больше в химической экологии, важное значение имеет агрегатное состояние вещества. Раньше считали, что существует три агрегатных состояния: твердое, жидкое и газообразное. Не так давно добавилось четвертое состояние вещества – плазма.
Любое вещество в зависимости от температуры и давления может находиться в том или ином агрегатном состоянии. Как правило, при низких температурах и высоких давлениях вещество находится в твердом агрегатном состоянии, а при высоких температурах и низких давлениях – в газообразном состоянии. При температурах порядка тысяч и миллионов градусов вещество переходит в ионизированный газ – плазму.
При обычных условиях – комнатной температуре и атмосферном давлении – химическое вещество находится в определенном для него, привычном для нас, стандартном агрегатном состоянии, например, Н2
О – жидкость, СО2
– газ, СаСО3
– твердое.
Знание особенностей каждого агрегатного состояния вещества необходимо не только химику, но и химику – экологу для понимания механизма процессов взаимодействия веществ.
Нахождение вещества в определенном агрегатном состоянии зависит как от природы, так и от характера взаимодействия частиц (молекул, атомов, ионов), образующих вещество. Следует иметь ввиду, что в обычных условиях атомы и молекулы практически теряют свою индивидуальность: вступая во взаимодействие, дают более высокую организацию вещества, чем индивидуальная молекула, образуя совокупность, названную агрегатным состоянием.
Переход от атомов и молекул к агрегатному состоянию вещества – химический процесс. Природа сил, обуславливающая образование агрегатного состояния, такая же, как и природа химической связи – электростатическая. Хотя переход из одного агрегатного состояния к другому не приводит к изменениям стехиометрического состава вещества, но он связан с определенным изменением его структуры. И поэтому данный процесс относится к химическому. Условие перехода из одного агрегатного состояния вещества в другое зависит от характера связи между частицами. Межагрегатный переход может сопровождаться изменением силового типа связи. Каждое агрегатное состояние характеризуется определенным характером движения частиц относительно друг друга и расстоянием между частицами. Так, если расстояние между частицами в твердом веществе порядка размеров самих частиц, то расстояние между частицами вещества в газообразном состоянии значительно превышают их размеры. Промежуточное положение занимают жидкости.
5.1. Твердое состояние.
Твердое состояние вещества является наиболее устойчивым агрегатным состоянием. Твердые тела характеризуются самостоятельной геометрической формой. Они обладают большим сопротивлением сдвигу, растяжению и сжатию. Все это обусловлено внутреннем строением твердого тела. В твердом теле частицы очень прочно связаны друг с другом, их средняя потенциальная энергия намного превышает среднюю кинетическую энергию частиц. Движение частиц в твердом теле очень ограничено, частицы могут совершать лишь незначительные колебания, не приводящие к изменению формы твердого тела.
Различают два вида твердого состояния вещества: кристаллическое и аморфное. Для кристаллического состояния вещества имеется строго упорядоченное расположение частиц и анизотропность свойств, т.е. неодинаковость его механических, электрических и других свойств по различным направлениям в пространстве. Аморфные тела не имеют строгого упорядоченного расположения частиц, их можно уподобить жидкостям с большой вязкостью. Характерной особенностью аморфных тел является одинаковое значение (изотропность) свойств при измерении в равных направлениях.
5.1.1. Кристаллическое состояние.
Подавляющее большинство твердых веществ имеют ту или иную кристаллическую структуру. Каждой кристаллической структуре соответствует своя геометрическая форма расположения частиц в пространстве. Кристаллом вещество обычно называют трехмерное его образование, характеризующееся строгой повторяемостью одного и того же элемента структуры (элементарной ячейки) во всех направлениях. Правильная форма кристалла обусловлена упорядоченным расположением составляющих его частиц – атомов, молекул или ионов. Кристаллическая решетка представляет собой пространственный каркас, образованный пересекающимися линиями соединяющими центры тяжести частиц – узлы решетки (рис.5.1.)

в
g
б b Рис.5.1. Кристаллическая решетка.
а a (черные кружочки – узлы решетки).
Каждый маленький объем жирный кубик на рис.5.1., являющийся наименьшим объемом кристалла, назван элементарной ячейкой кристалла. Элементарная ячейка любой формы кристалла содержит определенное число частиц вещества и характеризуется параметром ячейки – длиной ребра (а, б, в) и значениями углов между ними (a, b, g).
В зависимости от природы частиц, образующих кристалл, и характера связи между ними существует четыре типа кристаллических решеток – атомная, ионная, молекулярная и металлическая (рис.5.2.)
  + – + – + – + – + – + – + – + –
+ – + – + – + –
+ – + – + – + –
Рис.5.2. Типы кристаллических решеток: а) –атомная; б) –ионная;
в) –молекулярная; г) –металлическая.
а). Атомные решетки.
В узлах атомной решетки располагаются (находятся) нейтральные атомы. Связь между атомами осуществляется за счет ковалентных сил. Ковалентные связи многовалентных атомов в их соединениях имеют вполне определенную пространственную ориентацию. Та же ориентация атомов сохраняется и в кристаллической решетке. Таким образом, атомный кристалл можно рассматривать как гигантскую молекулу, все атомы которой связаны ковалентно.
Типичной атомной решеткой является решетка алмаза, состоящей из атомов углерода. Углерод 4-х валентен и его валентные электронные облака ориентированы в пространстве так что образуют между собой углы 1090
28’’. Каждый атом углерода находится в центре тетраэдра, вершины которого заняты другими четырьмя атомами углерода (рис.5.3.)

Рис. 5.3. Элемент кристаллической
решетки алмаза.
Такая тетраэдрическая структура, которая показана на рис.5.3. распространена по всему кристаллу. Ковалентные силы, действующие в атомных решетках очень велики и по этому такие решетки отличаются большой компактностью. Вещества с атомными решетками близкими к решетке алмаза имеют высокую твердость, высокую температуру плавления и малую летучесть.
Решетки типа алмаза имеет не только углерод, а и другие элементы четвертой группы – кремний, германий, серая модификация олова и ряд бинарных соединений, например, сульфиды (ZnS, CdS и др.)
б) Ионные решетки.
Ионная решетка характеризуется наличием в узлах пространственной решетки отдельных ионов.
Ионные решетки образуются правильным чередованием противоположно заряженных ионов, связанных между собой электростатическими силами притяжения разноименных зарядов. (см. рис.5.2. – б).
Каждый ион, входящий в решетку, находится в совершенно одинаковом отношении ко всем непосредственно окружающим его ионам противоположного знака. Молекулы ионных соединений в результате образования кристаллической решетки теряют свою индивидуальность, весь кристалл ионного соединения представляет собой единую макрочастицу.
Действующие в ионных структурах кулоновские силы обуславливают прочную связь между частицами. Поэтому для ионных кристаллов и температура плавления и твердость значительны. В качестве примера рассмотрим кристаллическую решетку хлорида натрия.

Рис.5.4. Кристаллическая решетка
хлорида натрия. –ионы Na+
, –ионы Cl–
.
d
Узлы решетки заняты ионами Na+
и Cl–
, причем каждый ион Na+
окружен 6-тью ионами Cl–
, а каждый ион Cl–
окружен 6-тью ионами Na+
. (рис.5.4.)
Как видно из рис.5.4., в кристалле NaCl каждый ион Na+
находится в центре октаэдра, шесть вершин которого заняты ионами хлора, так же и ион Cl–
окружен октаэдром из шести ионов Na+
.
Ионы удерживаются в решетке электростатическими силами. Расстояние между центрами ионов Na+
и Cl–
d=2,814 А0
.
По ионному принципу построены решетки почти всех солей многих оксидов и других соединений (MgO, PbS, CdS и др.).
Решетки такого типа, как у NaCl имеют все галогениды щелочных металлов (за исключением бромида и иодида цезия). Решетки типа NaCl отличаются друг от друга только межионными расстояниями d. В отличие NaCl, решетка CsJ – объемоцентрированный куб. В таком кристалле каждый ион окружен 8-мью ионами противоположного знака.
Для количественной характеристики окружения используют понятие “координационное число” (кч). Координационным числом данного атома (иона) называют число ближайших соседей в решетке без учета природы связи между ними. Так координационное число ионов Na+
в решетке NaCl равно 6. Аналогично координационное число ионов Cl–
равно тоже 6. В решетке CsJ координационное число ионов Cs+
и J–
равно 8.
Важной характеристикой кристаллической решетки является энергия решетки.
Энергией кристаллической решетки (Екр.
) называют работу, которую необходимо затратить на разрушение решетки и удаление ее составных частей на расстояние, при котором прекращается взаимодействие частиц. Ее относят к одному молю вещества и выражают в кДж/моль.
Энергию кристаллической решетки численно можно получить по формуле Борна.
 Екр
=К 3
Ör/М ; кДж/моль. Екр
=К 3
Ör/М ; кДж/моль.
здесь М – молекулярная масса твердого вещества; r – плотность; К – коэффициент.
Коэффициент К изменяется с изменением типа решетки. (Для решеток типа NaCl К=545, для решеток типа CsCl К=512, для решеток соединений типа ABrK>1500.)
Так [NaCl]n
=[Na+
]n
+ [Cl–
]n
; Екр
=773 кДж/моль.
в). Молекулярные решетки.
Молекулярные решетки образуются молекулами. Связь между молекулами осуществляется поляризационными ван–дер–вальсовыми силами. (рис.5.2. –в).
Так как ван–дер–вальсовы силы значительно слабее, чем электростатические или ковалентные, то и соединения с молекулярными кристаллическими решетками менее твердые, характеризуются малой прочностью, более летучи и имеют сравнительно низкие температуры плавления.
Отсутствие свободных ионов в кристаллах с молекулярными решетками объясняет малую растворимость в воде и очень малую электропроводность. Типичными представителями веществ с молекулярной решеткой являются многочисленные органические вещества.
По молекулярному типу построены решетки “замороженных” инертных газов. Все инертные газы (за исключением гелия), кристаллизуются в гранецентрированные кубические молекулярные решетки, а гелий – в плотную гексагональную упаковку.
Кроме инертных газов молекулярные кристаллы при затвердевании образуют и такие органические вещества, как H2
, N2
, O2
, P4
, S8
, H2
O, NH3
, HCl, SO2
, SiF4
и др.
г). Металлические решетки.
Металлическая решетка характерна для всех металлов в их твердом и жидком агрегатном состояниях. В узлах металлической решетки могут одновременно содержаться как нейтральные атомы, так и положительно заряженные ионы. Между узлами решетки свободно перемещаются электроны.
Так как все атомы данного металла одинаковы, каждый из них имеет равные с другими шансы на ионизацию. Иначе говоря, переход электрона от нейтрального атома к ионизированному может происходить без затраты энергии. Как следствие этого, в металлической структуре непрерывно осуществляется подобный обмен электронами и всегда имеется некоторое число электронов свободных, т.е. не принадлежащих в данный момент каким-либо определенным атомам.
Ничтожно малые размеры электронов позволяют им более или менее свободно перемещаться по всему металлическому кристаллу. Такой кристалл можно в связи с этим рассматривать как пространственную решетку из положительно заряженных ионов и нейтральных атомов, находящихся в атмосфере “электронного газа”.
Наличие свободных электронов во всех металлических структурах обуславливает существование общих свойств металлов. Сюда относят прежде всего такие характерные для них внешние признаки, как непрозрачность, металлический блеск и большей частью серый цвет. Со свободой перемещения электронов связана высокая электропроводность металлов и их хорошая теплопроводность.
Все эти особенности отличают металлы от других твердых веществ, в частности от веществ с атомной и ионной структурами.
Структурные типы химических соединений.
Расстояния между центрами атомов, молекул или ионов в кристаллических структурах не всегда одинаковы по длине. По характеру межцентровых расстояний в кристаллах различают следующие структурные типы: островные, слоистые, цепные и координационные. Соответственно этим структурным типам такое же название носят и кристаллические решетки.
 Островные решетки.
К островным решеткам относят прежде всего ионные решетки, в узлах которых находятся сложные ( комплексные) ионы, чередующиеся с обычными ионами. Так, в кристаллических комплексах, например [Ni(NH3
)6
]Cl2
или Na2
[SiF6
], островками, расположенными в узлах решетки, выступают октаэдрические комплексные ионы [Ni(NH3
)6
]2+
и [SiF6
]2–
. (рис.4.5.) Островные решетки.
К островным решеткам относят прежде всего ионные решетки, в узлах которых находятся сложные ( комплексные) ионы, чередующиеся с обычными ионами. Так, в кристаллических комплексах, например [Ni(NH3
)6
]Cl2
или Na2
[SiF6
], островками, расположенными в узлах решетки, выступают октаэдрические комплексные ионы [Ni(NH3
)6
]2+
и [SiF6
]2–
. (рис.4.5.)
Рис.5.5. Островная кристаллическая
структура комплекса К2
[SiF6
].
–ионы [SiF6
]2–
, –ионы натрия.
Слоистые решетки.
Слоистыми могут бать как атомные, так и ионные решетки. Основной идеей слоистых решеток является то, что длина связей между атомами (ионами), расположенными в одной плоскости короче и их прочность больше, чем расстояние между атомами, находящимися в разных плоскостях. Это значит, что связь между атомами, находящимися в одной плоскости имеет ковалентный характер (т.е. прочная). А связь между плоскостями близка по энергии ван–дер–вальсовым силам.
Примером слоистой атомной решетки является решетка графита (одной из модификаций углерода).
Атомы углерода занимают вершины правильных плоских шестиугольников со стороной 1,42А0
. Каждый атом углерода окружен тремя соседними атомами, с которыми связан ковалентными силами, образующими углы 1200
.
 1,42А0 1,42А0
3,4А0
Рис.5.6. Слоистая решетка графита.
Расстояние между параллельными плоскостями кристалла равно 3,4А0
. Атомы различных плоскостей притягиваются ван–дер–вальсовыми силами.
Такая структура обеспечивает мягкость графита и его удивительную слойность, что позволяет применять графит для карандашей и в качестве смазочного материала (графитовой смазки).
Цепочные структуры.
Как известно, молекулярные решетки имеют место в органической химии. В молекулярных решетках связь между молекулами осуществляется ван–дер–вальсовыми силами.
Значительный интерес представляют волокнистые строения ряда растительных и животных продуктов (целлюлоза, шелк, мускулы, нервные ткани и др.), сообщающие изделиям из таких продуктов своеобразные механические свойства (ткани, нити, веревки, бумага и т.д.).
Эти волокна образованы нитями, связаны ван–дер–вальсовыми силами, а сами нити состоят из длинной цепочки, образованной правильным чередованием атомных групп, связанных между собой ковалентными силами. Такие цепочки можно рассматривать как гигантские линейные молекулы.
Глава 8.
Энергетика химических процессов.
Любой химический процесс сопровождается тем или иным энергетическим эффектом: выделение или поглощение теплоты, света, выполнением электрической или механической работы.
Знание энергетических эффектов химических реакций необходимы не только химикам.
Раздел химии, в котором изучаются энергетические эффекты химических реакций, их зависимость от химического состава, строения и состояния веществ от условий проведения процессов, называется термохимией. Термохимия является составной частью химической термодинамики – области физической химии, в которой на основе законов общей термодинамики изучаются тепловые балансы химических реакций в различных условиях, устанавливается возможность и направление химических процессов. С помощью химической термодинамики выводятся законы химического и фазового равновесия и смещение этих равновесий в зависимости от изменения параметров состояния: температуры, давления, концентрации и др.
Для более глубокого понимания химической термодинамики необходимы рассмотреть ряд основных понятий этой науки.
8.1. Основные понятия и определения.
· Термодинамическая система.
Объект исследования в термодинамике называется системой. Термодинамической системой может быть любой макро***ческий объект (тело или группа тел, находящихся во взаимодействии, клоба с раствором вещества, штатив с набором химических реактивов и т.п.) выделенный из окружающей среды с помощью реально существующей или воображаемой поверхности раздела.

Термодинамическая система – комплекс взаимодействующих между собой физических тел реально или мысленно обособленный от окружающей среды
поверхность раздела
 окружающая термодинамическая окружающая окружающая термодинамическая окружающая
среда система среда
поверхность раздела
Для системы физическая граница не обязательна. Систему можно мысленно обосабливать от окружающей среды. Система может состоять из однородных частей, одинаковым по физическим и химическим свойствам и не однородных.
· Фаза.
Совокупность всех однородных частей системы, одинаковых по физическим и химическим свойствам и ограниченных от других частей системы поверхностью раздела называют Фазой
Различают системы однофазовые (гомогенные) и многофазовые (гетерогенные).
· Гомогенная система
– система состоящая из одной фазы. Это однородная система. Интенсивные свойства такой системы одинаковы во всех ее частях.
Гомогенной может быть система, состоящая из газообразных веществ. Жидкофазная система тоже может быть гомогенной.
· Гетерогенная система
– система состоящая из двух или более фаз. Между фазами имеется поверхность (граница) раздела. Это не однородная система. Хотя бы одно интенсивное свойство (например вязкость) изменяется скачком.
Примерами гетерогенной системы могут быть две несмешиваемые жидкости, находящиеся в колбе или кусочек металла, опущенный в раствор соляной кислоты.
Термодинамическая система, соприкасаясь с окружающей средой, может через поверхность раздела обмениваться с ней энергией и веществом или не обмениваться. В этом отношении различают изолированные системы и неизолированные.
· Изолированные системы
– это системы, у которых через поверхность раздела не может происходить обмен с внешней средой ни энергией, ни веществом.
Т.е. изолированной называют систему, имеющую, в частности, постоянный объем и лишенную возможности обмениваться с окружающей средой как веществом, так и энергией. Например: колба с водой, закрытая пробкой и температура воды такая, как окружающей среды. (Система будет изолированной).
· Неизолированные системы
могут быть закрытыми и открытыми.
¾ закрытые неизолированные
– такие системы, в которых через поверхность раздела может проходить обмен с внешней средой только энергией. Веществом система не обменивается.
¾ открытые неизолированные
– такие системы, в которых через поверхность раздела происходит обмен с внешней средой и веществом и энергией.

Термодинамическая система
гомогенная гетерогенная
изолированная неизолированная
закрытая открытая
Состояние системы определяет совокупность ее химических и физических свойств и описывается с помощью ряда переменных величин – параметров состояния (Р –давление, m –масса, Т –температура, С –концентрация, Е –энергия и т.д.). С помощью параметров состояния можно вывести другие переменные величины, которые называют термодинамическими функциями. (U –внутренняя энергия, Н –энтальпия, S –энтропия, G –энергия Гиббса, F –энергия Гельмгольца).
Это значит, что любая термодинамическая система, характеризуется с одной стороны параметрами состояния, а с другой – термодинамическими функциями.
характеризуется характеризуется
  Параметрами Термодинамическая Термодинамическими Параметрами Термодинамическая Термодинамическими
состояния система функциями
Р –давление U –внутренняя энергия
Т –температура Н –энтальпия
V –объем S –энтропия
m –масса G –энергия Гиббса
С –концентрация F –энергия Гельмгольца
Е –энергия
· Параметры состояния
– термодинамические параметры – независимые термодинамические переменные: P, T, V и т.д.
· Термодинамические функции
– функции состояния – величины, зависящие от термодинамических параметров состояния и не зависящие от пути перехода системы из одного состояния в другое (U, H, S, G, F).
Если система находится при Т–const, то она является изотермической, при Р–const – система изобарная, при V–const – система изохорная. Если две величины постоянные (Т–const и Р–const) – система является изобарно-изотермической.
Соответственно и процессы:
¾ Изитермические – процессы, протекающие при постоянной температуре.
¾ Изобарные – процессы, происходящие при постоянном давлении.
¾ Изохорные – при постоянном объеме.
8.2. Энергетические эффекты химических процессов.
В химических процессах чаще всего происходит выделение или поглощение теплоты.
· Количество теплоты, выделенной или поглощенной системой в результате химического превращения, называют тепловым эффектом
реакции.
Химические уравнения, в которых указано количество выделенной или поглощенной теплоты, называют термохимическими уравнениями.
В термохимических уравнениях указываются фазовые (агрегатные) состояния как исходных, так и продуктов реакции: г – газообразное, т – твердое, к – кристаллическое состояние.
J2
(к)
+H2
S(г)
=2HJ(г)
+S(к)
.
В таких уравнениях допускаются так же дробные коэффициенты.
½N2
(г)
+ ½O2
(г)
=NO(г)
.
SO2
(г)
+ ½O2
(г)
= SO3
(г)
.
· Если реакция протекает с выделением теплоты, то такую реакцию называют экзотермической
, а с поглощением теплоты эндотермической
.
Полная энергия системы состоит из трех видов энергии: кинетической энергии движения системы как целого объекта, потенциальной энергии обусловленной поглощением системы в каком-либо поле (гравитационном, магнитном, электрическом) и внутренней энергии системы.
Химические процессы, как правило, протекают в относительно стандартных условиях, т.е. при отсутствии электрических, магнитных и гравитационных воздействий. В этом случае изменение кинетической и потенциальной энергии системы практически не происходит. Все энергетические эффекты обусловлены только изменением внутренней энергии системы.
Внутренняя энергия системы
(U) включает в себя кинетическую и потенциальную энергию составляющих систему частиц. Это энергия взаимного расположения и движения молекул вещества, атомов входящих в состав молекулы, электронов, ядер и других частиц.
Измерить абсолютное значение внутренней энергии системы невозможно, но можно измерять изменение внутренней энергии ΔU в конкретном процессе, в частности в ходе химической реакции.
При переходе системы из начального состояния (1), от исходных веществ, в конечное состояние (2), к продуктам реакции, изменение внутренней энергии будет равно: ΔU =U2
–U1
8.3. Первый закон термодинамики.
В основе химической термодинамики лежат два закона, называемых первым и вторым законами термодинамики.
Первый закон термодинамики вытекает из обобщения многолетнего опыта человечества. Выдвинутые Ломоносовым идеи о законе сохранении материи и движения получили развитие в работах Майера, Гельмгольца и Джоуля, в которых экспериментально было установлено, что теплота и работа являются эквивалентными энергетическими эффектами и связаны с изменением внутренней энергии системы.
Первый закон термодинамики связан с законом сохранения энергии и устанавливает эквивалентность различных ее форм.
Первый закон термодинамики имеет следующую формулировку: Энергия, сообщенная системе, расходуется на увеличение (изменение) внутренней энергии и на работу, совершаемую системой против внешних сил
Математически первый закон термодинамики можно записать так:
 Q=ΔU +A Q=ΔU +A
Здесь: Q – энергия (теплота), сообщенная системе; ΔU – изменение внутренней энергии системы; А – работа против внешних сил.
Значение внутренней энергии системы зависит от параметров состояния системы (прежде всего от температуры и давления), а ΔU – от значения этих параметров в начальном и конечном состояниях системы. Следовательно, внутренняя энергия является термодинамической функцией состояния системы.
 ΔU = U2
–U1 ΔU = U2
–U1
U1
– внутренняя энергия системы в начальном состоянии. U2
– внутренняя энергия системы в конечном состоянии.
В обычных условиях система находится под атмосферным давлением, которое, не меняется резко. Его можно считать в данный момент постоянным. В этом случае работа будет совершаться за счет изменения объема, т.е. расширения или сжатия системы в результате химической реакции.
  А=V
1
∫V
2
pdv или А= pΔV=(V2
–V1
) А=V
1
∫V
2
pdv или А= pΔV=(V2
–V1
)
Значения ΔU и А подставим в математическое выражения первого закона термодинамики.
Q=ΔU + A=U2
– U1
+ p (V2
–V1
)=U2
– U1
+ pV2
– pV1
=(U2
+ pV2
) – (U1
+ pV1
)
Выражение (U + pV) обозначим через Н.
 U+ pV=Н U+ pV=Н
Следовательно
Q=H2
– H1
=ΔH
Величину Н называют энтальпией системы, а ΔH – изменением энтальпии системы в результате химической реакции. Мы пришли к выводу, что энергия (теплота), сообщенная системе, расходуется на изменение энтальпии системы. При Р const
 Qp
=ΔH Qp
=ΔH
Энтальпия Н, как и внутренняя энергия U является термодинамческой функцией, функцией состояния.
Рассмотрим, в чем заключается физический смысл энтальпии. В выражении
Н=U=pV
U – внутренняя энергия, а произведение pV – внешняя энергия. Следовательно энтропия – сумма внутренней и внешней энергии. Физический смысл энтальпии тот же, что и внутренней энергии, т.е. смысл энергии. Внутренняя энергия при постоянном объеме, энтальпия при постоянном давлении.
ПриР=const
Qp
=ΔH
При V=const
Q=ΔU
Это значит, что при постоянном давлении теплота процесса (тепловой эффект) равна изменению энтальпии, а при постоянном объеме теплота процесса равна изменению внутренней энергии.
Энтальпия – термодинамическая функция, определяющая энергию, необходимую для приведения данной системы в данное состояние, при этом учитывается изменение внутренней энергии и совершаемую работу
 Первому закону термодинамики можно дать и такую формулировку: Изменение внутренней энергии закрытой системы определяется количеством переданной теплоты и совершенной работы, т.е. Первому закону термодинамики можно дать и такую формулировку: Изменение внутренней энергии закрытой системы определяется количеством переданной теплоты и совершенной работы, т.е.
ΔU=Q–A
Выражение ΔU означает, что значение U, как функции состояния системы, не зависит от способа (пути) перехода системы из исходного состояния в конечное, а определяется только самим состоянием системы в исходном и конечном пунктах: ΔU=U2
–U1
. В тоже время теплота (Q) и работа (А) функциями состояния не являются, они возникают только в процессе перехода системы из первого состояния во второе и, естественно, зависят как от пути процесса, так и от условий его проведения. Разность (Q–A) дает ΔU не зависимо от способа перехода системы и определяет только приращение внутренней энергии системы, но не ее абсолютное значение.
Первый закон термодинамики объединяет три энергетические величины: внутреннюю энергию, теплоту и работу. Все величины Q, U, H и А имеют размерность энергии. В международной системе единиц (СИ) они выражаются в одних и тех же единицах – Джоулях (или Килоджоулях).
В связи с этим:
· Теплота
– это результат изменения внутренней энергии, это передача хаотического поступательного, колебательного и вращательного движения от структурных единиц системы к частицам внешней среды путем теплопроводности, излучения или конвекции (или наоборот).
· Работа
тоже является результатом изменения внутренней энергии системы. Это передача упорядоченного поступательного движения от организованного потока частиц системы к частицам внешней среды. С созданием в ней такого же организованного поступательного движения потока частиц. В частности работа расширения или сжатия системы за счет изменения объема в результате химического процесса.
Следовательно, работа является одной из форм передачи энергии от одной системы к другой – от системы совершающей работу к системе над которой работа совершается. При этом, энергия системы, которая совершает работу, будет убывать.
В экзотермических процессах система теряет тепловую энергию, поэтому энтальпия этого процесса со знаком минус: ΔHэкзот.реакц.
<0. Например,
Н2
(г)
+ ½О2
(г)
=Н2
О(г)
; ΔH0
= -57 кДж/моль.
В эндотермических процессах, наоборот, система приобретает энергию, следовательно, энтальпия идет со знаком плюс. (энергия вливается в систему).
SO3
(г)
= SO2
(г)
+ ½O2
(г)
; ΔH0
= +23,49 кДж/моль.
Так как значение ΔH зависит от параметров состояния, систему необходимо привести к стандартному состоянию.
· За стандартное состояние принимают наиболее устойчивое
состояние вещества при давлении 1атм и определенной постоянной температуре. Температура может быть любой, но приняли 298 К.
Различают три стандартных состояния соответственно трем агрегатным состояниям вещества.
Для твердых веществ наиболее устойчивым состоянием при давлении 1атм и тем-ре 298 К является кристаллическое, которое и принимается за стандартное.
Для растворенных веществ и ионов за стандартное состояние принимают состояние при мольности 1моль/кг; при этом предполагается, что раствор обладает свойствами бесконечно разбавленного раствора.
Если вещество при стандартных условиях может существовать в нескольких аллотропных формах, за стандартное принимают наиболее устойчивое состояние.
Изменение энтальпии в стандартном состоянии системы отмечают верхним индексом “0”: ΔH0
298К
.
Различают изменение энтальпии химического процесса и энтальпии образования вещества.
· За стандартную энтальпию образования вещества принимают стандартную энтальпию такой реакции, в которой 1моль этого вещества образуется из простых веществ, каждое из которых находится в термодинамически устойчивом состоянии.
Ее обозначим ΔH0
f
(298),СО2
= -396 кДж/моль, т.е.
С(графит)
+ O2
(г)
=СО2
(г)
; ΔH0
f
(298)
= -396 кДж/моль
Энтальпия образования простых веществ, в стандартном состоянии принимается равной нулю. Например ΔH0
f
(298),О2
=0; ΔH0
f
(298),Н2
=0; ΔH0
f
(298),С
u
2
=0 и т.д.
Стандартные энтальпии образования веществ величины табличные. По значениям стандартных энтальпий образования можно судить о устойчивости соединений. Чем более отрицательное значение энтальпии образования, тем более стойкое соединение. Например оксид ZnO устойчивее оксида CdO, так как ΔH0
f
(298),
ZnO
= -350,6 кДж/моль а ΔH0
f
(298),
CdO
= -260 кДж/моль.
8.4. Законы термохимии. Термохимические расчеты.
В основе термохимических расчетов лежит закон Гесса и его следствия.
В 1836г. Русский химик Гесс Г.И. экспериментально установил следующий закон:
· Тепловой эффект реакции (энтальпия реакции) при Vconst или Pconst не зависит от пути процесса (от числа промежуточных стадий), а определяется только начальным и конечным состоянием системы.
Наглядно этот закон можно показать на реакции образования СО2
.

  С +½О2
=СО СО + ½О2
= СО2 С +½О2
=СО СО + ½О2
= СО2
  ΔH0 ΔH0
 I
ΔH0
II I
ΔH0
II
С(гр.)
+ О2
СО2
(г.)
Исходное состояние ΔH0
х.р.
Конечное состояние
ΔH0
х.р.
=ΔH0
I
+ ΔH0
II
; -393,5= -1105 - 283
С(графит)
+ О2
= СО2
; ΔH0
= -393,5 кДж/моль
С(гр.)
+½О2
(г.)
=СО(г.)
; ΔH0
I
= -110,5 кДж/моль
СО(г.)
+ ½О2
(г.)
= СО2
(г.)
; ΔH0
II
= -283 кДж/моль
Закон Гесса позволяет рассчитывать энтальпии отдельных стадий, которых экспериментально определить трудно. Так, из рассмотренного примера можно рассчитать энтальпию реакции окисления СО до СО2
;
СО + ½О2
= СО2
, ΔH0
II
; ΔH0
II
= ΔH0
– ΔH0
I
= -393,5 – (-110,5)= -283 кДж/моль
Из закона Гесса вытекает ряд следствий. Некоторые из этих следствий раньше считались индивидуальными законами.
Рассмотрим два следствия из закона Гесса.
· Первое следствие.
Энтальпия процесса разложения вещества равна энтальпии образования, но имеет противоположный знак.
Так, энтальпия разложения СО2
на исходные элементы – графит и кислород равна энтальпии образования (-393,5 кДж/моль), но имеет знак плюс, т.е. ΔH0
разл. СО2
= 393,5 кДж/моль.
СО2
(г.)
= С(графит)
+ О2
(г.)
; ΔH0
= 393,5 кДж/моль
Это свойство можно сформулировать по другому:
· Тепловой эффект кругового процесса равен нулю.
 ΔH1 ΔH1
ΔH2
ΔH1
+ΔH2
=0
отсюда ΔH1
= -ΔH2
· Второе следствие.
Тепловой процесс реакции (энтальпия химического процесса) равен разности между суммой энтальпий образования конечных продуктов и суммой энтальпий образования исходных веществ.
ΔH0
реакции
= ∑ΔH0
f
(кон. прод.)
- ∑ΔH0
f
(исх. в-в.)
Пример.
Определить энтальпию реакции восстановления оксида меди (II) метаном при 250
С.
4СuO(к)
+СН4
(г)
=4Cu(к)
+CO2
(г)
+2H2
O(г)
.
Применим второе следствие из закона Гесса.
ΔH0
реакции
=4ΔH0
f
,
Cu
+ΔH0
f
,
CO
2
+2ΔH0
f
,
H
2
O
–[4ΔH0
f
,
CuO
+ΔH0
f
,
CH
4
].
Значения энтальпий образования всех продуктов данной реакции берем из таблицы стандартных энтальпий.
ΔH0
реакции
= 4*0+(- 393,8)+2(-242) - [4(- 162,1)+(-74,9)]= -154,7 кДж
ΔH0
реакции
= -154,7 кДж. Реакция экзотермическая.
Частные случаи применения закона Гесса.
Используя закон Гесса и его следствия можно, в частности, рассчитать энергию связи и энергию кристаллической решетки.
· Расчет энергии химической связи между атомами в молекулах.
Для того, чтобы найти энергию связи, необходимо, используя закон Гесса, рассчитать атомарную энтальпию (ΔH0
ат.
) энтальпию образования одного моля газообразного вещества из газообразных атомов. В качестве примера рассчитаем энергию связи О–Н в молекуле Н2
О.
По закону Гесса построим схему энтальпийной диаграммы для образования молекулы воды.
O(г)
 +2Н(г) +2Н(г)
ΔH0
дис.О2
2Н(г)
+ ½О2
(г.)
ΔH0
дис.Н2
Н2(г)
+ ½О2
(г.)
.
ΔH0
х.р.
Н2
О
ΔH0
х.р.
=ΔH0
дис.Н2
+ΔH0
дис.О2
+ΔH0
атом.
Значения энтальпий хим. реакции (ΔH0
х.р.
), диссоциации водорода (ΔH0
дис.Н2
), диссоциации кислорода (ΔH0
дис.О2
) получим используя второе следствие из закона Гесса. Значения энтальпий образования берется из соответствующих таблиц.
а) Н2(г)
+ ½О2
(г.)
=H2
O(г)
; ΔH0
х.р.
ΔH0
х.р.
=ΔH0
f
,
H
2
O
(г)
–[ΔH0
f
,
H
2(г)
+ ½ΔH0
f
,
O
2(г)
]= -241,84 – [0+½0]= -241,84 кДж/моль
б) Н2(г)
=2Н(г)
; ΔH0
дис.
H
2
ΔH0
дис.
H
2
=2ΔH0
f
,
H
(г)
– ΔH0
f
,
H
2(г)
=2*217,98 – 0= 435,96 кДж/моль
в) ½О2
(г.)
= O(г)
; ΔH0
дис.
O
2
=ΔH0
f
,
O
2(г)
– ½ΔH0
f
,
O
2(г)
= 246,8 – ½0=246,8
ΔH0
атом.
=ΔH0
х.р.
– ΔH0
дис.
H
2
– ΔH0
дис.
O
2
= -241,98 – 435,96 – 246,8 = -924,74 кДж/моль
Это значит,что в результате образования газообразной молекулы H2
O из газообразных атомов водорода и кислорода, т.е. в результате образования двух связей О–Н выделяется 924,74 кДж (ΔH0
атом.
= -924,74 кДж/моль).
В соответствии первого следствия из закона Гесса на разрушение молекулы воды (на разрушение двух связей О–Н) необходимо затратить 924,74 кДж/моль (ΔH0
разл.
= =+924,74 кДж/моль).
Следовательно, энергия одной связи О–Н будет равной: Ео-н
=924,74/2=462,37 кДж
· Расчет энергии кристаллической решетки.
Энергией кристаллической решетки называют то количество энергии, которое необходимо для разрыва кристаллической решетки на составные части с удалением их на значительное расстояние. Экспериментальное определение энергии кристаллической решетки затруднительно. Ее можно вычислить при помощи цикла Борна – Габера, основанного на законе Гесса, т.е. через определение энтальпии кристаллической решетки.
 Для определения энтальпии ионной кристаллической решетки хлорида натрия построим схему энтальпийной диаграммы. Для определения энтальпии ионной кристаллической решетки хлорида натрия построим схему энтальпийной диаграммы.
Na+
(г)
+Cl(г)
Na+
(г)
+Cl–
(г)
Na(г)
+Cl(г)
Na(г)
+½Cl2
Na(к)
+½Cl2
(г)
ΔH0
обр. NaCl
NaCl(к)
В этой схеме цикла Борна – Габера кристаллический хлорид натрия получают: с одной стороны непосредственно взаимодействием кристаллического натрия и газообразного хлора.
Na(к)
+½Cl2
(г)
= NaCl(к)
; ΔH0
обр. NaCl(г)
б) С другой стороны – посредством ряда стадий: испарения (сублимации) натрия - диссоциации хлора - ионизации (отрыва электрона) натрия - сродства к электрону хлора (присоединение электрона хлора) - взаимодействия иона натрия с ионом хлора с образованием кристаллического NaCl
· Na(к)
= Na(г)
; ΔH0
суб. Na
; ΔH0
суб. Na
=ΔH0
обр. Na(г)
– ΔH0
обр. Na(к)
· ½Cl2
(г)
= Cl(г)
; ΔH0
дис.
Cl2
; ΔH0
дис.
Cl2
=ΔH0
обр. С
l
(г)
–½ΔH0
обр. Cl2(г)
· Na(г)
= Na+
; ΔH0
ион.
Na
; ΔH0
ион.
Na
=ΔH0
обр. Na
+
(г)
– ΔH0
обр. Na
–
(г)
· Cl(г)
=Cl–
; ΔH0
ср.к эл.
Cl
; ΔH0
ср.к эл.
Cl
=ΔH0
обр. С
l
–
(г)
– ΔH0
обр. Cl(г)
· Na+
(г)
+ Cl–
(г)
= NaCl(к)
; ΔH0
реш
;
Используя закон Гесса, запишем.
ΔH0
обр. NaCl(к)
=ΔH0
суб. Na
+ΔH0
дис.
Cl2
+ΔH0
ион.
Na
+ΔH0
ср.к эл.
Cl
+ΔH0
реш
.
Все энтальпии, за исключением ΔH0
реш
, находят по следствии из закона Гесса.
ΔH0
обр. NaCl(к)
= -411 кДж/моль; ΔH0
суб. Na
=109 кДж/моль; ½ΔH0
дис.
Cl2
=121 кДж/моль; ΔH0
ион.
Na
=494 кДж/моль; ΔH0
ср.к эл.
Cl
= -380 кДж/моль.
Тогда:
ΔH0
реш
=ΔH0
обр. NaCl(к)
–ΔH0
суб. Na
–ΔH0
дис.
Cl2
–ΔH0
ион.
Na
–ΔH0
ср.к эл.
Cl
= -411 – 109 – 121– – 494 – (-380) = -755 кДж/моль.
Следовательно, энергия кристаллической решетки
Ереш.
= 755 кДж.
8.5. Энтропия. Энергия Гиббса. Направленность химических процессов.
Любая Термодинамическая система обладает не только определенным запасом внутренней энергии, но и характеризуется определенной степенью упорядочности.
 Существует специальная термодинамическая функция, которая характеризует меру хаотичности, беспорядка или, по другому, меру неупорядочности системы. Эту функцию называют энтропией
. Ее обозначают буквой “S”. Энтропия как мера неупорядочности системы является функцией состояния. Рассмотрим пример: Поместим в сосуд с перегородкой два газа азот и аргон. Обозначим это состояние системы S1
. В данном состоянии оба газа имеют определенную степень упорядочности Существует специальная термодинамическая функция, которая характеризует меру хаотичности, беспорядка или, по другому, меру неупорядочности системы. Эту функцию называют энтропией
. Ее обозначают буквой “S”. Энтропия как мера неупорядочности системы является функцией состояния. Рассмотрим пример: Поместим в сосуд с перегородкой два газа азот и аргон. Обозначим это состояние системы S1
. В данном состоянии оба газа имеют определенную степень упорядочности
N2
Ar (степень беспорядка). После того, как уберем перегородку, газы
S1
рис.8.1. начнут смешиваться. Это второе состояние системы обозначим S2
.
N2
Ar Степень беспорядка увеличилась. Произошло самопроизвольное
рис.8.2. увеличение энтропии ΔS=S2
–S1
.
· Cогласно второму закону термодинамики в изолированных системах энтропия самопроизвольно протекающего процесса возрастает. Это значит, что любая изолированная система, представленная самой себе, самопроизвольно изменяется в направлении максимальной хаотичности своего состояния. Энтропия является мерой хаотичности движения в системе, мерой молекулярного беспорядка.
Любое вещество состоит из частиц: молекул, атомов, ионов. В зависимости от агрегатного состояния свобода движения частиц различна. В веществе, находящимся в твердом состоянии “свобода” частиц ограничена, например, параметрами кристаллической решетки. Такая система достаточно высоко упорядоченная. Если взять жидкость, а тем более газообразное состояние, то система уже далека до упорядоченности. Чем тверже вещество, тем более оно упорядоченно, тем меньше для него значение энтропии. И, наоборот, уменьшение упорядоченности системы приводит к возрастанию ее энтропии.
Природа вещей такова, что частицам (молекулам, атомам, ионам,…) всегда присуще стремление к беспорядочному движению, в результате которого система стремится перейти из более упорядоченного состояния в менее упорядоченное. Возрастание степени беспорядка всегда влечет за собой возрастание энтропии (как функции беспорядка).
ΔS =Rln беспорядок в состоянии (II)/ беспорядок в состоянии (I)=Q/T
В конце прошлого века Больцман приписал энтропии статистический смысл.
S=KlnW
Здесь К – константа Больцмана К=R/N (R – газовая постоянная, N – число Авогадро), W – термодинамическая вероятность системы.
Термодинамическая вероятность системы равна числу микросостояний, которые необходимы для реализации данного макросостояния.
Единица измерения энтропии Дж/мольК. Энтропия растет с повышением температуры, при плавлении твердого вещества, при кипении жидкости, при переходе вещества из состояния с меньшей энергии в состояние с большей энергии и т.д.
В отличии от энтальпии для энтропии возможны экспериментальные определения абсолютных значений. Поэтому для стандартных энтропий образования обозначают не ΔS0
обр.
, а S0
обр
. Значком ΔS0
обозначают изменение энтропии в результате химического процесса.
Стандартная энтропия образования вещества – это абсолютная энтропия, соответствующая стандартному состоянию вещества при данной (стандартной температуре 298К). Обозначают ΔS0
298
.
Рассмотрим величину энтропии вещества при абсолютном нуле. Возмем правильно образованный кристалл любого чистого вещества (без примесей) и будем понижать температуру. Энтропия будет уменьшаться. При Т=0 число колебаний равно единице. Кристалл замерзает. При абсолютном нуле энтропия правильно образованного кристалла любого вещества в чистом виде( состоянии) равна нулю (S=0). Но если есть примеси или искажение ( деффекты) решетки, то энтропия не равна нулю(S≠0). Имеем остаточную энтропию S0
. При Т≠0, энтропия тоже не равна нулю. Отрицательных энтропий нет.
Изменение энтропии в химических реакциях вычисляют как разность между энтропиями конечного и начального состояний системы. Расчет ΔS0
х.р.
производится таким же приемом как и расчет ΔН0
х.р.
, т.е. по следствию из закона Гесса.
ΔS0
х.р.
= ΣS0
обр.(к.п.)
– ΣS0
обр.(ис. в-в.)
.
Энтальпия и энтропия отражают два противоположно направленных процесса любой системы. Если ΔН отражает в основном взаимодействие атомов в молекуле, стремление простых молекул к объединению в более крупные, т.е. стремление системы к состоянию с минимальным значением энергии, то ΔS отражает совсем противоположную тенденцию а именно, стремление к разрушению агрегатов и к беспорядочному расположению частиц.
Достижение системой минимальной энергии может быть только при ΔS=0. При ΔН=0 система самопроизвольно переходит в наиболее неупорядоченное состояние.
Стремление системы к минимальной энергии заставляет частицы взаимодействовать друг с другом и образовывать устойчивые агрегаты с наименьшим объемом, а тепловое движение расталкивает частицы увеличивая объем системы. В состоянии равновесия обе тенденции становятся равными, фактор энтальпии ΔН и фактор энтропии ΔS компенсирует друг друга.
 Поскольку ΔН измеряется в кДж/моль а ΔS измеряется в Дж/мольК, от для их количественного сопоставления необходимо привести к одинаковым единицам. Для этого ΔS умножают на Т. Получается равенство: Поскольку ΔН измеряется в кДж/моль а ΔS измеряется в Дж/мольК, от для их количественного сопоставления необходимо привести к одинаковым единицам. Для этого ΔS умножают на Т. Получается равенство:
ΔН=ТΔS
Если брать по отдельности, то химический процесс будет самопроизвольно протекать в сторону уменьшения общего запаса энергии системы и в сторону увеличения беспорядка в расположении отдельных частиц. Это две противоположно направленных тенденции любого химического процесса. Возникает вопрос, как их объединить и получить количественный критерий принципиальной осуществимости процесса. Критерий, с помощью которого можно определить, как далеко идет процесс, нельзя ли увеличить степень превращения исходных веществ в продукты реакции, как влияет на течение процесса температура, давление и другие факторы, можно ли заставить изучаемую реакцию протекать в обратном направлении. Такой критерий ввел в термодинамику американский ученый Гиббс в виде новой термодинамической функции, в последствии названной энергией Гиббса, которую обозначают буквой G. Для химического процесса ΔG. Величина ΔG связана с ΔН и ΔS следующим соотношением:
 ΔG=ΔН – ТΔS ΔG=ΔН – ТΔS
а) Если ΔG<0, то это есть условие возможности самопроизвольного протекания реакции в прямом направлении.
б) Если ΔG>0, протекание реакции в прямом направлении не возможно.
в) Если ΔG=0, наступает равновесие.
Следовательно, свободная энергия Гиббса является критерием протекания химического процесса. Мерой химического сродства является убыль G, т.е. –ΔG. Чем ΔG меньше нуля, тем дальше система от состояния химического равновесия, тем более она реакционноспособна. Величину ΔG называют “Свободной энергией” Гиббса. Что надо понимать под “Свободной энергией”? Свободная энергия – это часть внутренней энергии, которая может быть превращена в работу в данных условиях.
 –ΔG=Аmax
– pΔV=A’max –ΔG=Аmax
– pΔV=A’max
  Убыль энергии Гиббса в изотермическом процессе равна максимальной работе (Аmax
) за вычетом работы расширения (pΔV), т.е. максимально полезной работе (A’max
).Если из выражения G=H–TS найти TS=H–G , то получим разность (H–G). Эта разность между внутренней и свободной энергией называют “связанной энергией”. Она равна произведению энтропии на температуру. Связанная энергия – это та часть внутренней энергии, которая ни при каких условиях в работу превращена быть не может. Если из предыдущего выражения получим “S” Убыль энергии Гиббса в изотермическом процессе равна максимальной работе (Аmax
) за вычетом работы расширения (pΔV), т.е. максимально полезной работе (A’max
).Если из выражения G=H–TS найти TS=H–G , то получим разность (H–G). Эта разность между внутренней и свободной энергией называют “связанной энергией”. Она равна произведению энтропии на температуру. Связанная энергия – это та часть внутренней энергии, которая ни при каких условиях в работу превращена быть не может. Если из предыдущего выражения получим “S”
S=H–G/T
то можно сделать вывод, что энтропия равна доли связанной энергии (H–G) отнесенной к единице температуры.
О возможной направленности химического процесса можно судить по знакам изменения функций ΔН и ΔS. Влияние знака при ΔН и ΔS на направление протекания химического процесса представлено в следующей таблице.
Табл.8.1.
| Знак изменения функции |
Направление самопроизвольного протекания реакции |
| ΔН |
ΔS |
ΔG |
| – |
+ |
– |
Реакция протекает в прямом направлении при любых температурах. Она необратима |
+
|
–
|
+
|
В прямом направлении реакция невозможна ни при какой температуре. Она необратима. Может протекать только в обратном направлении. |
| – |
– |
± |
Реакция обратима. В прямом направлении реакция возможна при низких температурах. |
| + |
+ |
± |
Реакция обратима. В прямом направлении реакция возможна при высоких температурах. |
Если в результате расчета энергии Гиббса получится, что данная конкретная реакция при стандартной температуре (298К) не идет, необходимо выяснить ее обратимость, т.е. возможность процесса при других температурах.
При условии +ΔН и +ΔS реакция в прямом направлении возможна при высоких температурах. Для определении температуры реакции находим сначала температуру равновесия, а мы знаем, что условием равновесия ΔG=0. Тогда ΔН–ТΔS=0. Отсюда Трав.
= ΔН/ΔS.
Температура, при которой возможна реакция чуть больше температуры равновесия. Треакции
>Tрав.
Подобно стандартной энтальпии образования вещества ΔH0
обр.
в таблицах имеются значения стандартных энергий Гиббса образования веществ при стандартной температуре ΔG0
обр.298
. Эту величину можно рассчитать по известному уравнению:
ΔG0
298
=ΔН0
298
–298 ΔS0
298
.
Причем, ΔG0
298
образования простых веществ, аналогично ΔН0
298
образовании простых веществ, равны нулю.
Зная стандартные энергии Гиббса образования отдельных веществ можно по известному правилу (следствие из закона Гесса) рассчитывать энергии Гиббса конкретной реакции.
ΔG0
х.р.
=ΣΔG0
обр.(кон.прод.)
–ΣΔG0
обр.(исх.в-в)
Значения стандартных термодинамических функций образования веществ несут определенную информацию о этих соединениях. По величине стандартной энтальпии образования вещества (ΔН0
обр.
), ее знаке можно судить о прочности соединения. Так как ΔН0
обр.
характеризует энергию, которая выделяется (поглощается) в результате образования вещества из элементов, то, соответственно, для разрушения вещества на составные части (атомы) требуется такое же количество энергии, но взятой с противоположным знаком (следствие из закона Гесса). Большинство нейтральных (молекулярных) соединений имеют знак минус у энтальпий образования. Это значит, что они являются экзотермическими, обладающие меньшим запасом энергии, чем элементарные вещества, из которых они получены. И чем более отрицательная величина, тем более требуется энергии для разрушения молекулы на элементарные атомы. Эндотермическими являются некоторые группы соединений (гидриды, оксиды, нитриды, карбиды, металлы в газообразном состоянии, газообразные атомы неметаллов и небольшое число ионов в растворах). Для них ΔН0
обр.
имеет положительное значение. Это значит, что такие соединения, атомы, ионы получены с затратой энергии. Следовательно, такие состояния вещества является неустойчивым. Стандартная энтропия образования вещества S0
обр.
– всегда положительная величина, и чем больше ее численное значение, тем менее упорядочено вещество. По величине стандартной энтропии образования мы можем судить о агрегатном состоянии того или иного соединения, о наиболее устойчивой модификации одного и того же вещества, о разветвлении структуры молекулы и ряда других структурных особенностях химических соединений.
Остановимся на структурной энергии Гиббса образования вещества ΔG0
обр.
. Мы знаем, что ΔG является *** протекания химического процесса. Стандартная энергия Гиббса образования вещества является тоже энергией процесса, но процесса образования моля вещества из отдельных элементарных атомов, находящихся в наиболее устойчивом состоянии. В связи с этим, очевидно, что чем отрицательнее значение ΔG0
обр.
, тем устойчивее соединение. И, наоборот, чем более положительное значение ΔG0
обр.
, тем менее устойчивое вещество.
Рассмотрим несколько примеров.
1. Химическая инертность ряда соединений обусловлена большим отрицательным значением ΔG0
обр.
. Например, ΔG0
обр.
SiF
4
= -1572,5 кДж/моль. ΔG0
обр.
SF
6
= -1103,6 кДж/моль – достаточно инертные вещества.
2. Большая прочность комплексных соединений по сравнению с простыми тоже обусловлена значительным отрицательным значением энергии Гиббса их образования. ΔG0
обр.[
SiF
6]
2–
= -2134 кДж/моль. ΔG0
обр.[
BF
4]
–
= -1483 кДж/моль.
3. Для соединения Cl3
N значение ΔG0
обр.
величина положительная ΔG0
обр.
Cl
3
N
=293 кДж/моль. Это вещество неустойчиво, оно взрывоопасно.
8.6. Изменение термодинамических функций веществ, попавших в окружающую среду.
Поведение химических соединений попавших в окружающую среду значительно отличается от их поведения в небольших изолированных или закрытых системах. Здесь различие еще больше, чем отличаются между собой химические процессы в лабораторных опытах (в пробирках или колбах) и процессы в заводских условиях, в огромных реакторах и установках.
Экосистемы, водные и воздушные бассейны мы можем отнести к открытым системам. Эти системы способны изменять как свою массу, так и энергию. К большому сожалению, механизм процессов между антропогенными веществами (“загрязнителями” попавшими в окружающую среду), а также взаимодействие с элементами окружающей среды недостаточно изучен. Мы можем проводить только интуиктивные аналогии исходя из известных химических и химико-экологических закономерностей. В окружающую среду попадает не только огромное число соединений, разнообразных по химическим свойствам, но и большое количества тепла, выделяемого реакторами и промышленными установками.
Химическое, тепловое и радиационное загрязнение окружающей среды (природной термодинамической системы), приводит к резкому увеличению числа химических реакций, изменению скорости процессов и образованию сложных с заранее труднопредсказуемой структурой соединений.
В связи с этим на изменение термодинамических функций химических процессов будут влиять дополнительные факторы при относительном соблюдении общих тенденций.
Уравнение ΔG=ΔН – ТΔS объединяет два закона термодинамики и определяет принципиальную осуществимость химической реакции в любой термодинамической системе. Как следует из этого уравнения осуществление химической реакции зависит от двух факторов: энтальпийного и энтропийного. Согласно первого – каждая система стремится перейти в состояние с наименьшей внутренней энергией, выделив при этом в окружающую среду энергию в виде теплоты или работы или того и другого. По второму (энтропийному) фактору система стремится занять наиболее вероятное, наиболее неупорядоченное состояние (состояние с наиболее возможным значением энтропии).
Стремление системы к минимуму энергии приводит к тому, что любой химический загрязнитель окружающей среды будет стремиться к объединению с другими соединениями, образуя при этом новые, более сложные или комплексные соединения со специфическими свойствами, иногда относящихся к группе активных токсикантов.
Ярким примером может служить образование диоксинов в процессе хлорирования воды с целью ее обеззараживания.
Энтропийный фактор при взаимодействии веществ, попавших в окружающую среду, будет влиять на создание соединений с более разветвленной молекулярной конфигурацией, с нескольким функциональными активными группами, а следовательно, к образованию очень химико-биологически активных соединений.
И в том и в другом случае в естественных природных системах под действием антропогенных факторов образуются вещества-токсиканты, которые могут сильно повлиять на состояние экосистемы и привести к необратимым последствиям.
Глава 10.
Кинетика химических процессов.
10.1. Сущность кинетики химических процессов.
Химическая кинетика изучает как скорость, так и механизм химических реакций.
Под химической реакцией обычно понимают процессы превращения одних веществ в другие. В их основе лежат акты перемещения атомов от одних молекулярных структур к другим и изменение электронных состояний взаимодействующих частиц. Такие процессы возможны только при столкновении атомов или молекул взаимодействующих веществ. В этом смысле химической реакцией можно считать любое изменение вещества, при котором образуются или разрываются связи между атомами. При химической реакции происходит деление как энергии, так и атомов при сохранении общего числа атомов (соблюдение закона сохранения атомов).
В предыдущей главе, рассмотрели законы химической термодинамики, выяснили, что изменение свободной энергии Гиббса определяет принципиальную возможность проведения химической реакции (ΔG<0), и ΔG является “движущей силой” химического процесса. Однако эту величину нужно считать лишь необходимым
условием, но не достаточным для реального протекания процесса. В реальных условиях появляется ряд факторов вызывающий своеобразное “сопротивление” протеканию химической реакции.
Так, принципиально возможные химические процессы, не всегда осуществляются в действительности. Примером могут служить металлы (аллюминий, цинк, хром и др.), которые должны бы полностью окисляться кислородом воздуха, т.к. ΔG этих процессов меньше нуля, однако изготовленные из них детали и конструкции длительное время эксплуатируются в атмосфере воздуха. Аналогичное можно сказать о углеводородах, которые казалось бы самопроизвольно должны гореть на воздухе, но вопреки всем расчетам, они могут без изменения длительно пребывать в воздушной среде. Это объясняется тем, что процессы их окисления протекают очень медленно. В законах термодинамики фактор времени отсутствует. В реальных условиях химические превращения протекают во времени.
Многие химические реакции являются сложными, многостадийными процессами. Отдельные стадии реакции могут идти с большим трудом и этим обуславливать медленное протекание всего процесса. Образование отдельных промежуточных соединений может приводить к повышению энергии Гиббса эти стадии и являются своеобразным “барьером” на пути превращения исходных реагентов в продукты реакции.
При изучении хиических реакций важно знать не только почему протекает та или иная реакция, но и механизм, по которому происходит превращение и глубину этого превращения.
Под механизмом реакции обычно понимают сам процесс превращения, т.е. через какую стадию или ряд стадий должны пройти исходные вещества, чтобы превратиться в конечные продукты. Причем, каждая стадия для своего успешного завершения требует, чтобы произошли все предыдущие стадии.
Глубина превращения (степень превращения) характеризует насколько полно исходные вещества превращаются в продукты реакции. Из опытных данных ученые сделали вывод, что для протекания химической реакции необходимы столкновения молекул. Числом столкновений в секунду можно характеризовать скорость реакции. В газах и жидкостях столкновения происходят во всем объеме реакционной смеси, а в гетерогенных системах – на границе раздела фаз. Отсюда следует, что скорость химической реакции равна числу актов взаимодействия в единицу времени, в единице объема (для гомогенных реакций) или на единице поверхности раздела фаз (для гетерогенных реакций).
При каждом элементарном акте взаимодействия число молекул исходных веществ уменьшается, а число молекул продуктов реакции увеличивается. Это значит, что в результате химической реакции изменяются концентрации (или массы) как реагентов, так и образующихся веществ. На следующем рисунке (рис.10.1.) кривой “а” показано уменьшение концентрации исходного вещества, а кривой “б” –увеличение концентрации продукта реакции с течением времени”τ”.
 С С
моль/л а б
С1
С2
Рис.10.1.
τ1
τ2
τ
Скорость реакции количественно можно характеризовать изменением концентрации любого участвующего в реакции вещества за единицу времени. В этом случае концентрацию, как правило, выражают в моль, время – в секундах. Различают среднюю скорость реакции Vср
и мгновенную Vτ
.
Cредняя скорость
(Vср
) показывает изменение концентрации вещества (рис 10.1.) за определенный интрвал времени (от τ1
до τ2
). Она выражается следующим отношением:
Vср
= ± С2
– С1
/ τ2
– τ1
= ± ΔС/Δτ
Здесь знак “–“ относится к концентрациям исходных веществ он указывает на то, что концентрация исходных веществ убывает, а знак “+” относится к концентрациям продуктов реакции они в результате реакции возрастают.
Мгновенная скорость
Vτ
– это скорость реакции в данный момент времени τ, ее можно назвать истинной скоростью. Для того, чтобы определить мгновенную скорость в данный момент времени, необходимо определить изменение концентрации за бесконечно малый промежуток времени Vτ
=lim(-ΔС/Δτ). Мгновенная скорость мате-
Δτ–>0
матически определяется производной от концентрации по времени. Она равна тангенсу угла наклона касательной (углаα) к кривой, показывающей форму изменения концентраций от времени (на рис.10.3.) с течением времени. Величина угла наклона
 С С
k
α
Рис.10.3.
τ
касательной будет убывать, следовательно скорость реакции будет уменьшаться. Она рана тангенсу угла наклона (α) касательной к кривой зависимости концентрации от времени в соответствующий момент времени. (на рис.10.2. точка “k”)
С С
 α α
dc
dc k k
α
dτ τ dτ τ
Рис.10.2.
Vτ
=dc/dτ=tgα
Для реакции А+В=Д, VА
= -dCА
/dτ; VB
= -dCB
/dτ; VД
= +dCД
/dτ.
Если для определения скорости реакции брать: количество изменяющегося вещества – количество молей, единицу времени – секунды, а единицу реакционного пространства – литр (для гомогенных систем) и единицу площади межфазового пространства м2
(для гетерогенных систем), то
Vгомоген.
=[колич. вещества]/[время] [объем]=моль/с*л;
Vгетероген.
=[колич. вещества]/[время] [площадь]=моль/с*м2
.
Однак сокорость реакции можно определить по любому компоненту. Выбор вещества обуславливается легкостью, удобством и точностью определения количества вещества в реакционной системе. Например, объем выделеного газа, масса образующегося осадка, изменение кислотности раствора и др. На величину скорости химической реакции влияет множество факторов. Прежде всего это:
¾ природа реагирующих веществ;
¾ их концентраця;
¾ давление (если в реакции участвуют газы);
¾ катализаторы (ингибиторы);
¾ поверхности раздела фаз (для гетерогенных реакций);
¾ степень диффузии.
10.2.Факторы влияющие на скорость гомогенных реакций.
10.2.1. Химическая природа реагирующих веществ.
Для того, чтобы произошел химический процесс взаимодействия, частицам необходимо столкнуться. Столкнувшись, молекулы реагентов должны произвести рекомбинацию своих составных частей – атомов. Это значит, что в процессе столкновения должны разорваться химические связи в исходных молекулах и образоваться новые химические связи в молекулах продуктов реакции. Степень трудности разрушения химических связей в исходных реагентах зависит от энергии связи, а величина энергии связи является одной из основных качеств, характеризующих химическую природу вещества. Так скорость реакций
Н2
+F2
=2HF
Н2
+Cl2
=2HCl
будет разная, т.к. при всех одинаковых условиях энергия связи в молекуле фтора значительно меньше, чем в молекуле хлора. Следовательно, фтор будет легче распадаться на атомы и взаимодействовать с водородом. В понятие “природы реагирующих веществ” входит совокупность факторов, определяющих структуру и реакционную способность частиц. Ею определяется специфика взаимодействия. Так, реакции с участием полярных молекул протекают быстрее, чем процессы между неполярными молекулами. Известно также, что при взаимодействии молекул большое влияние на скорость оказывает расположение функциональных групп.
Превращение веществ происходит за счет перераспределения электронных плотностей между составляющими их атомами, что приводит к разрушению старых и образованию новых химических связей.
10.2.2. Концентрации взаимодействующих веществ.
Как уже отмечалось, взаимодействие между молекулами реагентов может происходить только при их контактах. Чем чаще происходят столкновения молекул, тем быстрее протекает реакция. Так как число столкновений зависит от концентрации, то с увеличением концентрации увеличивается и скорость реакции.
Форма зависимости скорости реакции от концентраций реагирующих веществ определяется так называемым законом действующих масс. Впервые закон действующих масс был сформулирован в прошлом веке (1864 – 1867гг) норвежским химиком К. Гульдбергом и П. Вааге. Сейчас известно, что этот закон справедлив только для элементарных реакций.
По сложности процесса взаимодействия реакции делятся на простые (элементарные) и сложные. Простые реакции протекают в одну стадию по стехиометрическому уравнению. Сложными являются реакции, протекающие через ряд последовательных стадий, параллельных направлений, цепные, сопряженные таких реакций большинство.
Закон действующих масс, справедливый для элементарных (простых) реакций, имеет следующую формулировку:
· Скорость элементарной химической реакции при данной температуре пропорциональна произведению концентраций реагирующих веществ в степенях с показателями, равными стехиометрическим коэффициентам в уравнении реакции.
Для реакции, записанной в общем виде аА+вВ=dD, скорость, согласно закону действующих масс, будет выражена следующим кинетическим уравнением
V=K[A]a
[B]в
.
Кинетическое уравнение – это форма зависимости скорости реакций от концентраций реагентов. В этом уравнении [А] и [В] – концентрации реагирующих веществ; k – константа скорости данной реакции. Физический смысл константы скорости заключается в том, k равна скорости V, если концентрации реагирующих веществ равны единице [A]a
=1 моль/л; [B]в
=1 моль/л или [A]a
[B]в
=1 моль/л. По-другому, константа скорости k – это удельная скорость реакции. Константа скорости зависит от природы реагирующих веществ, температуры, катализатора и площади поверхности раздела фаз (для гетерогенных реакций).
В зависимости от числа молекул (частиц), участвующих в элементарном химическом акте, различают молекулярность реакции.
Встречаются моно-, би- и тримолекулярные реакции. В их элементарном акте участвуют соответственно одна, две и три молекулы. Например;
а) N2
O5
=NO2
+NO+O2
–мономолекулярная реакция;
б) 2NO2
=N2
O4
–бимолекулярная реакция;
в) 2NO+Cl2
=2NOCl –тримолекулярная реакция.
Кинетические уравнения этих реакций имеют вид:
а) V=K[N2
O5
]
б) V=K[NO2
]2
в) V=K[NO]2
[Cl2
]
Элементарный акт взаимодействия более трех молекул (частиц) практически неизвестен. Поэтому четыре и более молекулярные реакции не встречаются.
Кроме молекулярности реакции в химической кинетике оперируют также понятием “порядок реакции”.
· Порядок реакции – это сумма показателей степеней при концентрациях веществ в кинетическом уравнении закона действующих масс.
Для реакций, приведенных выше, порядок реакции равен соответственно единица (реакция а), двум (реакция б) и трем (реакция в). Для сложных реакций “порядок реакции“ как правило, не равен сумме показтелей степени.
Для реакции:
аА+вВ+…=dD
сумма показателей степени будет
а+в+…=n
Порядок реакции здесь (η≠n) соответственно, и в кинетическом уравнении показатели степеней у концентраций реагентов не равны а, в, … . В этом случае кинетическое уравнение нужно записать так:
V=K[A]α
[B]β
(где α≠а‚ β≠в)
Порядок сложных реакций (в частности реакций, идущих через промежуточные стадии) определяется экспериментальным путем. Он может быть и дробной величиной. Дробными могут быть и показатели степеней α‚ β и т.д.
Например, для реакций
Н2
+Br2
=2HBr
в начальный период взаимодействия кинетическое уравнение будет иметь вид:
V=K[Н2
][Br2
]0,5
Порядок реакции n=1+0,5=1,5
Размерность константы
скорости К зависит от суммы показателей степени при концентрациях веществ, т.е., от порядка реакции.
В реакциях первого порядка V=K[A] размерность К будет с –1
(K=V/[A]=моль/л*с/моль/л=1/с).
В реакциях второго порядка V=K[A]2
размерность К равна л/моль*с.
В реакциях 3-го порядка V=K[A]3
константа К имеет размерность л2
/моль2
*с.
В реакциях n-го порядка константа К будет имеет размерность л(
n–1)
/моль(
n–1)
*с.
Скорость сложных химических процессов, протекающих через ряд промежуточных реакций обычно определяют по той промежуточной реакции, которая идет наиболее медленно.
10.2.3. Влияние температуры.
Скорость реакции сильно зависит от температуры. Можно привести такой пример: реакция между водородом и кислородом (т.е. реакция окисления водорода 2Н2
+ О2
=2Н2
О) при обычных условиях практически неосуществима. При температуре 318 0
С она произойдет за 230 лет, но при температуре 700 0
С происходит взрыв, т.к. скорость реакции очень высока, реакция осуществится за 0,007 сек.
Обобщая экспериментальные данные голландский ученый Вант–Гофф в 1879 году
· установил следующее правило: повышение температуры на каждые 10 градусов увеличивает скорость гомогенных реакций в 2–4 раза.
Число, показывающее во сколько раз увеличивается скорость реакции при повышении температуры на 10 градусов, названо температурным коэффициентом скорости. Температурный коэффициент скорости (коэффициент Вант–Гоффа) обозначают буквой γ.
 Vt2
= Vt1
* γ(t2
–t1
/10) уравнение Вант–Гоффа Vt2
= Vt1
* γ(t2
–t1
/10) уравнение Вант–Гоффа
где Vt2
и Vt1
–скорости реакции соответственно при температурах t2
и t1
(t2
>t1
); γ – температурный коэффициент.
γ=К(t+10)/Kt
=V(t+10)/Vt
=2–4
Kt
1
/Kt
2
= γt
2–
t
1/10
Для реакции окисления водорода повышение температуры от 273 до 3730
при γ=2 скорость увеличивается в 1024 раза.
V373
/V273
= γ373–273/10
=2100/10
=210
=1024
Уравнение Вант–Гоффа является упрощенным выражением зависимости скорости реакции от температуры. С его помощью можно лишь примерно оценить влияние температуры на скорость реакции. Более точная зависимость скорости от температуры представлена соотношением шведского ученого Сванте Аррениуса (1889г)
 Аррениус показал, что константа скорости реакции К имеет следующую зависимость от температуры Аррениус показал, что константа скорости реакции К имеет следующую зависимость от температуры
lnK= -(E/RT+C)
отсюда К=А*е-(
E/
RT)
. В этом выражении А – постоянный множитель, не зависящий от температуры и концентраци; е – основание натурального логарифма (равное 2,713); Т – абсолютная температура в Кельвинах (К); Е – энергия активации (кДж/моль), т.е. та энергия, которую нужно сообщить молекулам (частицам), находящимся в исходном состоянии, чтобы они могли вступить в реакцию; R – универсальная газовая постоянная (8,314 Дж/моль*К).
По теории Аррениуса химическая реакция может происходить только при столкновении активных частиц
. Активными считаются частицы, имеющие такое количество энергии, которое необходимо для осуществления реакции, т.е. для преодоления сил отталкивания, возникающих между электронными оболочками частиц, и их взаимодействия. Активные частицы, прежде чем превратиться в конечный продукт, при столкновении друг с другом образуют промежуточную группировку, называемую активированным комплексом. Активированный комплекс является очень неустойчивым переходным состоянием системы, в котором происходит перераспределение химических связей. В дальнейшем исходные химические связи окончательно разрушаются и образуются конечные связи, образуются продукты реакции.
Для реакции
А2
+В2
=2АВ
процесс взаимодействия графически можно изобразить так:
  А В А … А А А А В А … А А А
  | + | | + | | + | | + |
А В В … В В В
начальное состоя- переходное состоя- конечное состояние
ние системы ние системы (акти- системы (продукты
(реагенты) вированный комплекс) реакции)
Для того, чтобы исходные вещесива (реагенты) достигли переходного состояния (превратились в активированный комплекс) система должна получить определенное количество энергии, называемое энергией активизации.
Энергия активизации
, входящая в уравнение Аррениуса, представляет собой разность между средней энергией реагирующих частиц и энергией активированного комплекса.
 Энергия активизации – своя величина для каждого процесса, это значит, что она является характеристической величиной конкретной реакции можно сказать, что она определяет влияние на скорость химической реакции природы реагирующих веществ. Если сравнивать энергию активизации и энергию диссоциации веществ, то энергия активизации по величине меньше, чем энергия диссоциации наименее прочной связи в молекулах реагирующих веществ. На следующей энергетической схеме для экзотермической реакции А+В=АВ (рис.10.4.) дано соотношение между величиной различных энергий. Энергия активизации – своя величина для каждого процесса, это значит, что она является характеристической величиной конкретной реакции можно сказать, что она определяет влияние на скорость химической реакции природы реагирующих веществ. Если сравнивать энергию активизации и энергию диссоциации веществ, то энергия активизации по величине меньше, чем энергия диссоциации наименее прочной связи в молекулах реагирующих веществ. На следующей энергетической схеме для экзотермической реакции А+В=АВ (рис.10.4.) дано соотношение между величиной различных энергий.
Активированый
комплекс
Н А…В
Е
А+В
ΔН реакции
Σ Н реагентов
Σ Н продукты реакции
путь реакции
Рис.10.4. Энергетическая схема хода реакции А+В=Д
Активация молекул может быть осуществлена при нагревании или растворении вещества, при выделении энергии в ходе самой реакции, при поглощении ими квантов светового, радиоактивного, рентгеновского или другого излучения, под действием ультрозвука, электрического разряда и даже при ударе о стенку сосуда.
 По теории Аррениуса зависимость ln”K” от 1/Т имеет линейный характер (рис.10.5.) По теории Аррениуса зависимость ln”K” от 1/Т имеет линейный характер (рис.10.5.)
lnK
lnK2
Рис.10.5. Зависимость lnK от 1/Т
lnK1
α
0
1/T1
1/T2
1/T
Исходя из графической зависимости lnK от 1/Т можно расчитать энергию активации.
Е=Rtgα
Это значит, что энергию активации можно определять из анализа экспериментальных данных по зависимости скорости реакции от температуры.
По закону действующих масс скорость реакции А+В=АВ выражается следующим кинетическим уравнением:
V=K[A][B]
Выражение К определяем из соотношения Аррениуса:
К=А*е–Е/
RT
и подставляем в кинетическое уравнение реакции:
V= А*е–Е/
RT
[а][в]
Произведем замену величины А[а][в] на Z и получаем значение:
V=Z*е–Е/
RT
Прологарифмировав это выражение, будем иметь уравнение:
lgV=lgZ – (E/RT)lge=lgZ – (0,434E/R)*1/T
lgV= -(0,434E/R)*1/T
которое является уравнением прямой в координатах lgV и 1/T, как это представлено на рис.10.5.
 lgV lgV
lgZ Рис.10.5.
β
1/T
Из графика (рис.10.5.) определяют тангенс угла β:
tgβ=lgV/(1/T) = ((-0,434E/R)*(1/T))/1/T = -0,434E/R
Последнее выражение позволяет определить энергию активации Е
Е= -tgβR/0,434
10.2.4. Влияние давления.
В случае взаимодействия газообразных реагентов, на скорость реакции влияет также давление . Повышение давления равноценно увеличению концентрации газов. Сжатие системы в два раза соответственно приводит к увеличению концентрации каждого из газов тоже в два раза. Мы уже знаем, что зависимость скоростьи простых реакций от концентраций определяется законом действующих масс. Следовательно, по этому закону можно в принципе оценивать, во сколько раз изменится скорость реакции при изменении величины давления в данной системе.
В рамках теории химической кинетики зависимость константы скорости от давления выражается следующим уравнением:
(σlnK/Pσ)T= -ΔV/RT
в котором ΔV – разность между суммой мольных объемов исходных веществ и мольным объемом активированного комплекса, т.е. изменение объема при переходе реагентов в активированное состояние.
10.3. Катализаторы. Гомогенный катализ.
Скорость реакции часто зависит от присутствия в системе “постороннего” вещества с которым реагенты способны образовывать промежуточные соединения (активированный комплекс) и этим ускорять реакцию. Такие “посторонние” вещества называют катализаторами.
Катализатором называют такое вещество, которое своим присутствием и участием в реакции изменяет скорость, но в конечном итоге выделяется в первоначальном (качественном и количественом) виде. Увеличение скорости реакции при помощи катализатора называют катализом. Если катализатор находится в таком же агрегатном состоянии что и реагенты и между взаимодействующими веществами и катализатором нет поверхности раздела, то такой катализ называют гомогенным.
Сущность и механизм гомогенного катализа.
 Исходные вещества (реагенты) превращаясь в продукты реакции должны преодолеть энергетический барьер, равный энергии активизации. Еа
(рис.10.6.) Исходные вещества (реагенты) превращаясь в продукты реакции должны преодолеть энергетический барьер, равный энергии активизации. Еа
(рис.10.6.)
Н А…В
ΔЕкат.
Еа
А…К АК…В
Еа1
Еа2
А+В А+В
К(
kat
)
AK
ΣН исх. реаг.
АВ АВ+К
ΣН кон.прод.
Путь реакции
Рис.10.6. Энергетическая диаграма а) реакции А+В=АВ без катализатора;
б) каталитической реакции А+В+К=АВ+К
Еа
– энергия активации реакции без катализатора;
Еа1
и Еа2
– энергия реакции каталитической реакции;
АК – соединение вещества катализатора с одним из реагентов;
А…К, АК…В – активированные комплексы каталитической реакции;
А…В – активированный комплекс обычной (некатализируемой) реакции;
ΔЕкат.
– снижение энергии активизации под действием катализатора.
Сущность гомогенного катализа заключается в том, что катализаторы уменьшают величину энергетического барьера. Это происходит по следующему механизму: катализатор взаимодействует с одним из реагентов, образуя промежуточный комплекс. Затем этот промежуточный (активированный) комплекс взаимодействует с вторым реагентом, образуя конечные продукты и высвобождая катализатор в неизменном первоначальном виде.
За счет образования промежуточного комплекса реагент-катализатор энергтический барьер уменьшается на величину ΔЕкат.
. Примером гомогенного катализа может служить реакция окисления SO2
в SO3
в нитрозном способе получения серной кислоты.
2SO2
+O2
+NO(
кат
.)
=2SO3
+NO
Без катализатора реакция идет медленно и процесс неэффективный. Катализатор, оксид азота (II), первоначально взаимодействует с кислородом, образуя активированный комплекс NO2
.
2NO+O2
=2NO2
Затем это промежуточное соединение легко взаимодействует с оксидом серы (IV), окисляя его до SO3
и выделяя в первоначальном виде катализатор.
NO2
+SO2
=SO3
+NO
Применение катализатора NO сопровождается уменьшением энергетического барьера на величину ΔЕкат.
и значительно ускоряет реакцию.
10.4. Особенности кинетики гетерогенных реакций.
Гетерогенные реакции – это реакции между химическими реагентами, находящимися в различных агрегатных состояниях. Таких процессов очень много. К ним относятся горение топлив, взаимодействие металлов с кислотами, получение азотной кислоты абсорбцией оксидов азота водой, выщепачивание кислотами руд, обработка нефтепродуктов серной кислотой и др.
Особенностью гетерогенных процессов является то, что взаимодействие между реагентами происходит на границе раздела фаз. На скорость таких реакций влияют как химические так и физические факторы. К последним относятся величина поверхности раздела фаз и быстрота переноса вещества из объема к границе раздела и от нее в объем. Для увеличения поверхности раздела фаз необходимо твердое вещество измельчать и распылять один из двух несмешивающихся жидких реагентов. Следовательно, на скорость гетерогенных реакций влияет степень дисперсности реагента.
Так как твердое вещество в результате взаимодействия изменяет только свою массу (концентрация его всегда постоянна), то в кинетическое уравнение закона действующих масс твердое вещество не включается.
Как уже было сказано, химическая реакция в гетерогенных системах протекает на поверхности раздела фаз. Для того, чтобы непрерывно протекала реакция необходима постоянная доставка реагента к поверхности раздела фаз и уноса с нее уже образовавшегося вещества. Как видим, процесс делится на три последовательные стадии: диффузия реагента в зону взаимодействия, химическая реакция, удаление продукта реакции.
В соответствии с теорией диффузии: диффузионный поток тем интенсивнее, чем большн разность между концентрацией реагента в данной точке объема (Со
) и в зоне реакции (Ср
), больше коэффициент диффузии Д и меньше тощина слоя (δ), через который происходит массопередача. В случае стационарного режима (т.е. режима, при котором за рассматриваемый промежуток времени на реакцию расходуется все вещество, доставленное к поверхности раздела фаз), скорость реакции может быть расчитана по следующему уравнению, связывающему скорость процесса с химическим (К) и диффузионным (Д/δ=β) факторами.
V=(K*β/K+β)*Со
Здесь встречается два случая:
1. Медленно протекает сама химическая реакция. (К мало, “химическое сопротивление” К–1
значительное. Процесс протекает в так называемой кинетической области. Для увеличения скорости необходимо применять теже способы воздействия на реакцию, как в гомогенных системах.
2. Медленным является сам процесс переноса вещества (велико “диффузионное сопротивление” β–1
). В этом случае для увеличения скорости применяют перемешивание.
Гетерогенный катализ.
Если взаимодействующие вещества и катализатор находятся в разных фазовых (агрегатных) состояниях, катализ – гетерогенный. В гетерогенных каталитических реакциях катализатором является твердое вещество. Например, платиновый катализатор используется при окислении аммиака, катализаторы на основе меди и золота – при синтезе высокомолекулярных соединений (пластмасс и смолы), цинка и хрома – в производстве метанола, ванадий – при получении серной кислоты и т.д.
В случае гетерогенного катализа взаимодействие между реагентами протекает на поверхности катализатора. Механизм процесса состоит из 5-и стадий.
Первая стадия – диффузия реагентов к катализатору.
Вторая стадия – адсорбция реагентов на поверхности катализатора. (на этой стадии происходят изменения в электронном строении реагентов и снижается энергетический барьер).
Третья стадия – реакция на поверхности катализатора.
Четвертая стадия – десорбция продуктов реакции.
Пятая стадия – диффузия продуктов в объем.
Ускорение процесса при гетерогенном катализе, как и в гомогенном, объясняется образованием активированного комплекса. Для увеличения поверхности катализатора его стараются делать губчатым. На выступающих точках (вершинах) катализатора, называемых активными центрами не только адсорбцируются молекулы реагентов, но и претерпивают изменения, в результате которых облегчается образование конечных продуктов.
Рассмотрим этот процесс на примере синтеза аммиака:
N2
+H2
=2NH3
После адсорбции азота и водорода на поверхности твердого катализатора происходит разрыв связей между атомами азота в молекуле азота N≡N и между атомами водорода в молекуле водорода Н–Н. На эту операцию затрачивается энергия. Однако каждая разорвавшаяся связь в молекуле азота, так же и в молекуле водорода насыщается за счет образования связи с катализатором. Образуется промежуточный комплекс Kat–N и Kat–H. Происходит выделение энергии, чем частично компенсируется затрата энергии на разрыв связей в молекулах реагентов. На последующем этапе происходит разрыв связей в промежуточных комплексах Kat–N и Kat–H и образование молекул продуктов реакции NH3
. При этом выделяется значительная энергия и катализатор высвобождается для дальнейших актов взаимодействия.
Иногда для усиления эффективности катализатора применяют дополнительные вещества, называемые промоторами. Промоторы сами не являются катализаторами, но повышает активность катализаторов. Например, применяемый в производстве серной кислоты катализатор V2
O5
повышает свою активность в присутствии оксида бария или аллюминия.
За счет уменьшения энергии активации путем применения катализаторов скорость реакции возрастает во много раз. В следующей таблице 10.1. приведены значения энергии активации некоторых процессов без катализатора и с катализатором.
Таблица 10.1.
| Реакция |
Энергия активации Еа
кДж/моль |
катализатор |
| без катализатора |
с катализатором |
| С2
Н4
+Н2
=С2
Н6
|
180 |
40 |
платина |
| 2HJ=H2
+J2
|
200 |
60 |
платина |
| 2SO2
+O2
=2SO3
|
250 |
60 |
платина |
| 2NH3
=N2
+3H2
|
326 |
167 |
железо |
| 2H2
O2
=2H2
O+O2
|
750 |
55 |
иод |
Используя уравнение:
К=Ас-Еo/
RT
можно оценить, во сколько раз увеличится скорость реакции при каталитическом уменьшении эенргии активации. Например, если энергию активации снизить с 251 до 167 кДж/моль то скорость реакции возрастает в е20
раз.
Другие факторы, влияющие на скорость.
а) Растворитель.
Влияние растворителя обусловлено многими факторами – ван-днр-вальсовыми и дисперсионными взаимодействием, электростатич. взаимодействием между ионами и диполями, сольватацией и др.
б) Электрический разряд.
В этом случае скорость реакции пропорциональна мощности электрического разряда.
в) Радиационное воздействие.
В результате прохождения ионизирующего излучения через вещество.
г) Фотохимическое воздействие.
–под действием света.
Если постороннее вещество замедляет реакцию, то такой отрицательный катализатор называется ингибитором.
Например, реакция разложения Н2
О2
замедляет глицирин. Следовательно глицирин является ингибитором Н2
О2
.
10.5. Цепные реакции.
Одной из разновидностей класса сложных реакций являются цепные реакции. Если для других типов реакций скорость с течением времени уменьшается, так как уменьшается концентрация реагентов, то у цепных реакций наоборот, увеличение скорости со временем.
Цепной реакцией называют химическое взаимодействие реагентов, в котором первоначально появившаяся активная частица (возбужденный атом или радикал) приводит не к одному, а к множеству превращений, и передают свою энергию возбуждения вновь образовавшимся частицами.
Появление первоначальной активной частицы (возбужденного атома или радикала) может произойти в результате любого энергетического импульса (кванта света, электрического разряда, электронного удара, местного повышения температуры). Каждая активная частица вызывает целую цепь последующих превращений и резко увеличивает скорость химического взаимодействия. Так смесь водорода с хлором при комнатной температуре на рассеяном свету практически не взаимодействует. Но как только такую смесь осветить прямым солнечным светом, то она начинает активно реагировать и может произойти даже взрыв.
Существует два типа цепных реакций, реакций с неразветвляющимися и с разветвляющимися цепьями. Примером первого типа цепных реакций может служить процесс синтеза хлорида водорода из молекулярного водорода и молекулярного хлора. При освещении смеси газообразных хлора и водорода под действием кванта света молекула хлора распадается на две активных частицы. Происходит зарождение цепи.
Cl2
+hv=2Cl* (звездочкой отмечена активная частица)
Далее активный хлор Cl* приводит в действие механизм развития цепи.
Cl*+H2
=HCl+H*
H*+Cl2
=HCl+Cl
Cl*+H2
=HCl+H*
и так далее
Каждая молекула активного хлора (частица Cl*) может привести к образованию до 104
(100000) молекул хлорида водорода. Реакция между хлором и водородом представляет собой длинную цепь последовательно протекающих элементарных процессов.
Обрыв цепи возможен при столкновении двух одинаковых частиц.
Cl*+Cl*=Cl2
; H*+H*=H2
.
Однако вероятность такого процесса мала, так как образование малекул из атомов сопровождается выделением энергии, которая вновь приводит к разрыву образующихся связей. И процесс взаимодействия между водородом и хлором идет до конца. Чтобы осуществить обрыв цепи необходимо осуществить отвод энергии. Это возможно с помощью твердого тела: частицы примеси, стенки сосуда и др.
Реакция с разветвляющимися цепьями отличаются от реакций с неразветвляющимися цепьями тем, что возникновение одного сводного радикала может привести к образованию сразу нескольких активных частиц и процесс начинает развиваться лавинообразно.Примером реакции с разветвляющейся цепью может служить окисление водорода кислородом. Начало цепи даст при определенных условиях реакция:
Н2
+О2
=ОН*; ОН*+Н2
=Н2
О+Н* или Н2
+hv=2H*
Далее идет развитие и разветвление цепи.
Н2
О
ОН*+Н2
ОН*…
 Н*+О2 Н*+О2
Н*+О2
О*…
Н2
О
ОН*+Н2
 О*+Н2
Н*… О*+Н2
Н*…
ОН*…
Н*+О2
О*…
Такой механизм очень типичный для реакций идущих со взрывом. Увеличение активных центров определяется “коэффициентом размножения”. Если этот коэффициент больше единицы (1,1–1,2), то скорость реакции непрерывно нарастает и процесс переходит в фазу взрыва.
Механизм цепных реакций очень сложный. На развитие реакции влияет скорость зарождения активных частиц, скорость разветвления цепи, скорость ее обрыва, а также ряд внешних факторов – давление, температура, скорость отвода тепла.
Разработка теории цепных реакций начата Боденшнейном (1913). Однако математическая теория и физические основы течения цепных реакций заложены и развиты в работах Н.Н.Семенова, Н.М.Эммануэля, Хиншельвуда. Разработанная ими теория цепных процессов получила широкое применение в современной технике и энергетике.
Глава 11.
Химическое равновесие.
11.1. Причины обратимости химических процессов.
Самопроизвольно протекающие химические реакции можно разделить на две группы: необратимые и обратимые.
Необратимые реакции протекают только в одном направлении. В этих реакцииях исходные вещества (реагенты) практически полностью превращаются в стехиометрическом состоянии в продукты реакции. Необратимости реакции способствуют условия, при которых хотя бы один из продуктов реакции уходит из реакционной зоны в виде осадка, газообразного вещества или представлять собой малодиссоциирующее в реакционной среде вещество.
Примером необратимых реакций могут быть следующие взаимодействия:
AgNO3
+NaCl=AgCl↓+NaNO3
Na2
CO3
+2HCl=CO2
↑+NaCl+H2
O
Соблюдается такое правило: чем менее растворимым является продукт реакции, чем труднее диссоциирует труднодиссоциируемое соединение, тем полнее протекает необратимая реакция. Необратимую реакцию нельзя повернуть в обратную сторону без ввода новых реагентов и без затраты энергии.
Существует множество реакций, которые не идут до полного превращения реагентов в продукты, взаимодействие как бы прекращается на определенном этапе. В реакционной смеси обнаруживаются как продукты реакции, так и исходные вещества. На самом деле реакция не прекращается, а только с определенного момента продукты реакции начинают взаимодействовать и выделять исходные вещества, т.е. начинает протекать обратная реакция. Такие реакции называются обратимыми. Обратимыми называются реакции, которые при данных условиях одновременно протекают в двух взаимно противоположных направлениях.
аА+вВ↔сС+dD
К обратимым относятся следующие реакции.
N2
+3H2
↔2NH3
H2
+J2
↔2HJ
2SO2
+O2
↔2SO3
В обратимых реакциях вместо знака равенства ставится взаимнонаправленные стрелки, указывающие на обратимость процесса.
Обратимые реакции характеризуются химическим равновесием.
Под химическим равновесием понимают не изменение во времени (при постоянных давлении, объеме и температуре) состояние системы, содержащей вещества, способные к химическому взаимодействию.
Различают истинное и кажущееся (метастабильное) равновесия.
Истинное химическое равновесие характеризуется тремя признаками:
¾ в системе не происходит видимых во времени изменений при отсутствии внешних воздействий;
¾ равновесие достигается как при прямой, так и при обратной реакции;
¾ самое малое внешнее воздействие легко смещает равновесие в ту или другую сторону.
Метастабильным (кажущимся) равновесием является таким состоянием системы, при котором из-за некоторых “тормозящих” факторов химическая реакция не доходит до состояния истинного равновесия. Торможение химической реакции может происходить как в самом начале процесса, так и в некоторый момент, если возникают тормозящие факторы. Метастабильное равновесие отличается от истинного тем, что при устранении “тормозящих” факторов, реакция идет до достижения истинного равновесия. Для истинного равновесия ΔG0
х.р.
=0, а для метастабильного ΔG0
х.р.
<0. Термодинамическим условием наступления истинного является ΔG0
х.р.
=0.
Хотя при химическом равновесии ΔG0
=0, но взаимодействие веществ не прекращается реакция продолжается. Молекулы реагентов двигаются, соударяются, образуют новые вещества которые от соударения снова распадаются на исходные вещества. Равновесное состояние – это такое состояние, при котором число образовавшихся молекул продукта реакции равно числу распавшихся молекул на исходные вещества.
Состояние химического равновесия любой равновесной системы сохраняется до тех пор, пока сохраняются в неизменном виде внешние факторы (температура, давление) и в систему не вводятся дополнительно никакие вещества (ни реагенты, ни продукты реакции).
Рассмотрим химическое равновесие с точки зрения закона действующих масс. Для обратимого процесса, изображенного в общем виде:
V
 аА+вВ↔сС+dD аА+вВ↔сС+dD
V
 Скорость прямой реакции (V) с течением времени уменьшается, а скорость обратной – увеличивается (V) (Рис.11.1.) Скорость прямой реакции (V) с течением времени уменьшается, а скорость обратной – увеличивается (V) (Рис.11.1.)
V
V
V=V
равновесие Рис.11.1.
V
время
Запишим кинетические уравнения прямой и обратной реакции.
V=K1
[A]a
[В]в
V=K2
[C]c
[D]d
Для состояния равновесия V=V
Приравняем правые части кинетических уравнений
K1
[A]a
[В]в
= K2
[C]c
[D]d
Берем отношения константы скоростей
K1
/K2
=[C]c
[D]d
/[A]a
[В]в
Заменим отношение постоянных величин констант скоростей K1
/K2
на постоянную величину К, называемую константой равновесия. Получим:
К=[C]c
[D]d
/[A]a
[В]в
Для конкретной равновесной системы
N2
(г)
+3Н2
(г)
↔2NH3
(г)
выражение константы равновесия будет следующим
К=[NH3
]2
/[N2
][Н2
]3
Для равновесных систем закон действующих масс может быть сформулировать так: Химическое равновесие устанавливается, когда произведение концентраций продуктов реакции, возведенных в степени, равные стехиометрическим коэффициентам, деленное на произведение концентраций реагентов, возведенных в соответствующие степени, становится постоянной величиной при определенных условиях.
Константа равновесия является количественной характеристикой химического равновесия. Она не зависит от начальных концентраций реагирующих веществ, но зависит от температуры. Константа равновесия не зависит также и от пути реакции, ее механизма, а определяется только значением равновесных концентраций реагентов и продуктов реакций.
Зная величину константы равновесия и исходные концентрации реагентов можно расчитать равновесные концентрации всех веществ.
Константа равновесия химических реакций связана со стандартным изменением энергии Гиббса этой реакции ΔG0
следующим уравнением.
ΔG0
= -2,3RTlgKT
При температуре 250
С (Т=298К)
ΔG0
298
= -5,69lgK298
(кДж/моль)
11.2. Факторы, влияющие на химическое равновесие.
Достижение истиного химического равновесия для химического процесса энергетически выгодно. (т.к. ΔG =0). Однако для промышленной технологии установление равновесия между продуктами реакции и реагентами экономически невыгодно, так как снижает выход конечного продукта. Это ставит перед химиками задачу смещения равновесия в сторону получения максимального количества продукта реакции. Такого эффекта можно добиться изменением условий, при которых установлено равновесие.
Экспериментально определено, что при изменении концентрацй веществ, давления в реакторе, температуры проведения процесса, изменяется скорость как прямой, так и обратной реакции. Равновесие в системе нарушается и происходит его смещение в сторону той реакции, скорость которой больше. Спустя некоторое время система снова приходит в состояние равновесия, но уже отвечающее новым (изменившимся) условиям.
Рассмотрим по-отдельности основные факторы влияющие на равновесие.
а). Изменение концентраций реагентов.
Для гомогенной равновесной системы аА+вВ↔сС+dD при неизменных давлении и температуре изменение концентрацй веществ приводит к смещению равновесия. Записываем выражение константы равновесия.
К=[C]c
[D]d
/[A]a
[B]в
как известно, константа равновесия при неизменной температуре –величина постоянная.
При увеличении концентрации исходных веществ (Реагентов “А” и “В”) равновесие должно сместиться вправо, т.е. в сторону увеличения концентраций продуктов реакции. К этому выводу мы придем, анализируя выражение константы равновесия. Так как “К” – величина постоянна, то при увеличении концентрации реагентов [А] и [В], стоящих в знаменателе, должен увеличиваться числитель, т.е. концентрации продуктов реакции [C] и [D]. Происходит дальнейшая реакция с получением дополнительного количества конечного продукта. При этом, естественно, уменьшается концентрация исходных веществ. Следовательно, увеличив концентрацию исходных веществ, мы смещаем равновесие в сторону прямой реакции, т.е. реакции расходующеей добавленное количество реагентов до установления нового равновесия.
Сместить равновесие вправо можно и путем вывода части получаемых продуктов из зоны реакции. В нашем случае – уменьшением концентрации веществ [С] с [D] равновесие смещается в сторону прямой реакции.
Изменение давления.
Изменение давления влияет на состояние равновесия систем, содержащих газообразные вещества. Изменение давления равноценно изменению концентрации всех газообразных веществ. Это значит, что в большей мере изменяется скорость той реакции, в которой участвует большее количество молекул газов.
Если в системе N2
(г)
+3Н2
(г)
↔2NH3
(г)
повысить давление в два раза, то в два раза увеличится концентрации каждого из веществ. Однако молекул исходных веществ больше, чем число молекул продукта реакции, то скорость прямой реакции будет выше скорости обратной реакции и поэтому равновесие смещается вправо.
Следовательно, можно сделать такой общий вывод: повышение давления смещает равновесие в сторону реакции, содержащей меньшее число молей газа.
N2
+ 3Н2
↔ 2NH3
 1моль 3моль 2моль 1моль 3моль 2моль
4моль 2моль
4объема 2объема
 направлени смещения равновесия направлени смещения равновесия
Чем больше изменение объема системы в прямой реакции, тем больше влияние давления на сдвиг равновесия, но если в процессе взаимодействия объем системы не
меняется, то изменение давления не влияет на равновесие. Так, в системе
Н2
(г)
+J2
(г)
↔2HJ изменение давления не смещает равновесие ибо до реакции и после объем не изменяется.
Изменение температуры.
Для выяснения влияния температуры на смещение равновесия конкретной равновесной системы необходимо знать энтальпию этой системы. Если прямая реакция эндотермическая, т.е. идет с поглощением теплоты (+ΔН), то обратная реакция будет экзотермической (–ΔН). При повышении температуры ускоряется как прямая, так и обратная реакции, но в разной степени. Для обратимых реакций энергия активации эндотермического процесса больше энергии активации экзотермического процесса. Чем больше энергии активации, тем сильнее скорость реакции зависит от температуры.
Следовательно, при увеличении температуры происходит смещение химического равновесия в сторону эндотермической реакции, т.к. в результате этой реакции поглощается теплота и система охлаждается.
Для системы:
аА+вВ=сС; +ΔН
повышение температуры Т↑ смещает равновесие вправо (→).
Изменение внешних условий, при которых система находится в равновесии, приводит к смещению равновесия в сторону реакции противодействующей вызванному изменению.
Это универсальное правило сформулировано французским химиком-технологом Ле Шателье и названо впоследствии принципом Ле Шателье.
· Если на систему, находящуюся в истинном химическом равновесии, воздействовать из вне путем изменения какого-либо параметра, влияющнго на равновесие (концентраця, давление, температура), то равновесие смещается в сторону той реакции, которая способствует востановлению первоначального состояния системы.
Влияние катализатора.
Катализатор равновесие не смещает, т.к. он не является ни реагентом, ни продуктом реакции. Катализатор в одинаковой степени изменяет скорость как прямой, так и обратной реакции. Этим способствует быстрейшему достижению химического равновесия, т.е. он обеспечивает достижение химического равновесия за меньший промежуток времени.
11.3. Особенности равновесия в гетерогенных системах.
Реакции между веществами, находящимися в различных агрегатных состояниях протекает на поверхности раздела фаз. Если в гетерогенной равновесной системе какое-то вещество находится в твердом состоянии, то добавление в систему этого вещества не приведет к смещению равновесия, т.к. концентрация этого вещества постоянна, независима от величины его массы.
Так, для следующей гетерогенной равновесной системы:
FeO(тв)
+H2
(г)
↔Fe(т)
+H2
O(г)
выражение константы равновесия не будет содержать ни оксида железа, ни железа:
К=[H2
O(г)
]/[H2
(г)
]
А для системы:
CO3
(
тв
)
↔CaO(
тв
)
+CO2
(
г
)
в выражение константы равновесия входит только СО2
: К=[СО2
]
11.4. Химико-экологические равновесия.
Химическая реакция в любой неживой или живой системе самопроизвольно протекает только в направлении ведущему к достижению равновесия. После достижения равновесия изменений в системе не наблюдается. Рассматриваемые нами ранее системы, системы лабораторных и промышленных масштабов, относятся к системам закрытого типа. В этих системах, как правило, может происходить обмен с окружающей средой только энергией, но за границы системы не происходит проникновение вещества.
Микро- и макро-экосистемы, являются открытыми системами, т.к. они обмениваются с окружающей средой как энергией, так и веществом. Существующее или возникающее равновесие в этих системах зачастую носит временной характер.
Глобальные природные или антропогенные воздействия на установившееся равновесие в микро- или макроэкосистеме приводит к смещению этого равновесия и протеканию химических или физикохимических процессов могущих привести к изменению самой структуры системы. Рассмотрим конкретный пример: эрозия горных, ландшафтных пород, протекающих под воздействием воды, углекислого газа и кислорода. Карбонатные составляющие этих пород вступают в реакцию с водой и углекислым газом.
CaCO3
+H2
O+CO2
↔Ca(HCO3
)2
Мы знаем, что подобные реакции идут медленно и достигают равновесия. Но в природе процесс ускоряется и равновесие смещается вправо. Поступающий с атмосферными осадками к карбонатным породам (растворимый в воде) углекислый газ и отвод в окружающую среду продукта реакции Ca(HCO3
)2
вызывает сдвиг равновесия и разрушению горных пород.
В экосистемах без антропогенного воздействия длительное время могут существовать псевдоравновесия, т.е. кажущиеся равновесия. Например, такие микроэкосистемы как сухая древесина, каменный уголь, торф, нефть из-за высокой энергии активации находятся в равновесии с окружающей средой.
Однако в результате нарушения этого псевдоравновесия загораются торфяники, залежы угля, газовые или нефтяные месторождения.
Реакции в открытых системах очень часто являются необратимыми (хотя в закрытых системах эти процессы являлись бы равновесными). Это обусловлено прежде всего тем, что часть конечного продукта удаляется, выводится из сферы взаимодействия и он уже не может участвовать в обратной реакции.
Даже такие микросистемы как растущее и гниющее дерево можно рассматривать с позиции открытых систем. Растущее дерево постоянно поглощает питательные вещества (химические соединения), углекислый газ, воду и энергию, этим процессом оно создает растительную ткань и выделяет кислород. Процесс односторонний – дерево растет.
Гниющее дерево, наоборот, поглощает кислород и выделяет углекислый газ, воду и энергию. Гниение – процесс, который идет до конца, т.к. продукты реакции удаляются, их концентрация никогда не достигает равновесного значения. Энергия рассеивается.
Глава 12.
Растворы.
12.1. Общая характеристика растворов. Классификация.
Если кто-то предполагает, что жидкофазное состояние вещества является раствором, допускает ошибку. Растворы бывают не только жидкими, но твердыми и газообразными. Отличительной особенностью раствора является то, что они состоят из двух и более веществ, причем эти вещества настолько перемешаны, что составляют гомогенные системы. В отличии от растворов жидкое состояние вещества содержит молекулы одного типа. (Дистиллированная вода – молекулы H2
O, толуол – молекулы С6
Н5
–СН3
и тд.)
 Раствором называется гомогенная система (гомогенная смесь), состоящая из двух и более компонентов, одним из которых является растворителем, а остальные – растворимые вещества. Схематично состав раствора можно представить так: Раствором называется гомогенная система (гомогенная смесь), состоящая из двух и более компонентов, одним из которых является растворителем, а остальные – растворимые вещества. Схематично состав раствора можно представить так:
 Раствор (гомогенная смесь) = Растворимое вещество + Растворитель Раствор (гомогенная смесь) = Растворимое вещество + Растворитель
компоненты раствора
Растворителем является то вещество, которое находится в таком же агрегатном состоянии, что и раствор. Типы растворов приведены в следующей таблице.
Таблица 12.1.
Тип
раствора
|
Компоненты раствора
(агрегатное состояние)
|
Примеры
растворов
|
| Растворитель |
Растворимое вещество |
| газовый* |
газ |
газ |
воздух |
жидкость
|
жидкость |
газ |
кислород в воде, соляная кислота |
| жидкость** |
жидкость |
спирт в воде |
| жидкость |
твердое |
соль в воде |
твердый***
|
твердый |
газ |
раствор водорода в платине |
| твердый |
жидкость |
ртуть в серебре (амальгама) |
| твердый |
твердый |
серебро в золоте |
Примечание к таблице 12.1.
* Газовые растворы обычно называют газовыми смесями.
** Если оба вещества являются жидкостями, то раствором выступает та жидкость, которой больше.
*** Твердые растворы бывают двух типов: растворы внедрения и растворы замещения. Растворы внедрения образуются в том случае, когда молекулы растворяемого вещества в два и более раза больше, чем молекулы растворителя. В этом случае молекулы внедряются в кристаллическую решетку растворителя и занимают место в пространстве между узлами решетки. Примером раствора внедрения является раствор водорода в платине. В растворах замещения атомы растворяемого вещества замещают часть атомов растворителя в узлах решетки. Такие растворы образуются в случае близости размеров атомов растворителя и вещества. Примером является раствор серебра в золоте.
В первом случае, когда растворителем является жидкость, в среде которой растворяемое вещество “раздробляется” до молекул (или атомов) и распределяется в ней в виде нейтральных частиц, образуются растворы неэлектролитов. Примером раствора неэлектролита может быть раствор сахара в воде. Во втором случае, когда в жидком растворителе, например в воде, растворяемое вещество, кроме обычного растворения, еще и распадается (диссоциирует) на ионы, которые распределяются в среде растворителя, образуются растворы электролитов. Примером тагого раствора электролита может быть раствор поваренной соли (NaCl) в воде.
Растворы электролитов проводят электрический ток, их называют проводниками второго рода, т.к. они обладают ионной проводимистью.
Вещества, не диссоциирующие на ионы под действием растворителя, называются неэлектролитами. К ним относится большое число органических соединений, содержащие неполярные или слабо полярные молекулы (сахар, крахмал, глюкоза). Растворы неэлектролитов не проводят электрический ток. Электролитами называют вещества, растворы или расплавы которых проводят электроток. Электролитами являются вещества с ионной и сильно полярными ковалентными связями.
12.2. Растворимость. Факторы влияющие на растворимость.
Растворимостью называется способность вещества растворятся в данном растворителе при данной температуре.
Различают:
¾ неограниченную растворимость (вода – спирт, жидкие KCl – KBr).
¾ ограниченная (вода – эфир, жидкие LiCl – KCl).
¾ практическое отсутствие растворимости (вода – керосин, жидкие LiF – CsCl).
Мерой растворимости является концентрация вещества (его содержание) в насыщенном растворе.
Растворы бывают: насыщенные, пересыщенные и ненасыщенные. Насыщенным считается раствор, который находится в равновесии с растворяемым веществом. В насыщенном растворе содержится предельное при данных условиях количество растворенного вещества. Раствор является пересыщенным, если в нем содержится растворенного вещества больше, чем расчитано по растворимости. Раствор, содержащий вещества меньше, чем определено по растворимости, называется ненасыщеным.
На практике растворимость вещества выражается величиной, называемой коэффициентом растворимости. Коэффициент растворимости показывает массу вещества, насыщаемую при данной температуре 100 граммов растворителя.
Растворимость некоторых веществ в воде представлена в таблице 12.2.
Таблица 12.2.
| Вещество |
Растворимость: масса вещества (г) в 100 г воды при температурах |
| 00
C |
100
C |
200
C |
400
C |
600
C |
800
C |
1000
C |
| SO2
|
22,83 |
16,21 |
11,29 |
5,41 |
3,2 |
2,1 |
– |
| NH3
|
89,7 |
68,3 |
52,9 |
31,6 |
16,8 |
6,5 |
0 |
| CuSO4
|
14,3 |
17,4 |
20,7 |
28,5 |
40,0 |
55,0 |
75,4 |
| K2
SO4
|
7,35 |
9,22 |
11,11 |
14,76 |
18,17 |
21,4 |
24,1 |
| Al2
(SO4
)3
|
31,2 |
33,5 |
36,4 |
45,7 |
59,2 |
73,1 |
89,0 |
| NaCl |
35,7 |
35,8 |
36,0 |
36,6 |
37,3 |
38,4 |
39,8 |
| NH4
Cl |
29,4 |
33,3 |
37,2 |
45,2 |
55,2 |
65,6 |
77,3 |
| KNO3
|
13,3 |
20,9 |
31,6 |
63,9 |
110,0 |
169 |
243 |
| KNO2
|
278,8 |
– |
298,4 |
334,9 |
350 |
376 |
412,9 |
Процесс растворения протекает в две стадии. На первой стадии происходит разрушение агрегатного состояния растворяемого вещества. На этой стадии происходит затрата энергии. Энтальпия первой стадии процесса растворения имеет знак плюс.
ΔHI
>0
На второй стадии процесса растворения происходит образование сольватов (в частном случае гидратов), т.е. группировок, состоящих из молекул (ионов) растворяемого вещества, окруженных молекулами растворителя. Процесс сольватации сопровождается выделением теплоты. Энтальпия второй стадии процесса растворения идет со знаком минус.
ΔHII
<0
В целом, процесс растворения будет эндотермическим (раствор будет охлаждатся), если на разрушение агрегатного состояния растворяемого вещества тратится больше энергии, чем ее выделяется в процессе сольватации. Такой эндоэффект (ΔHраств
>0) наблюдается при растворении твердых веществ. Например, растворение в воде Na2
S2
O3
*7H2
O приводит к сильному охлаждению раствора. И, наоборот, если растворяются газы или жидкости в жидких растворителях, то процесс сольватации выделяет значительно больше энергии, чем ее затрачено на первой стадии процесса (ΔHII
>ΔHI
) раствор сильно нагревается. Этот тепловой эффект процесса растворения называется экзоэффектом. (ΔHраств
<0).
На процесс растворения влияют следующие основные факторы (природа растворяемого вещества и растворителя, их агрегатное состояние, температура, давление, наличие в растворе посторонних веществ).
1. Природа растворяемого вещества и растворителя.
Существует классическое правило “подобное растворяется в подобном”. Полярные вещества (ионные соединения и соединения с полярной ковалентной связью) лучше растворяются в полярнрм растворителе. Для растворения неполярных веществ необходимо применять неполярные соединения.
2. Температура. Давление.
О влиянии температуры на растворимость можно говорить только в общих чертах, т.к. этот процесс не однозначный. Как правило, повышение температуры увеличивает растворимость тех веществ, процесс растворимости которых эндотермичный, т.е. идет с поглощением теплоты (ΔHраств
>0).
Рассмотрим несколько частных случаев.
а). Растворимость твердых веществ в жидкостях.
С повышением температуры растворимость твердых веществ увеличивается, так на разрушение кристаллической решетки твердого тела расходуется энергии больше, чем ее выделяется при сольватации (гидротации). ΔHI
>ΔHII
. Однако степент увеличения растворимости от температуры различна (табл.12.2.). В некоторых случаях кривая растворимости может проходить через своеобразный максимум. Примером может служить растворимость сульфата натрия в воде.
б). Растворимость газов.
При повышение температуры растворимость газа в жидком растворителе уменьшается, т.к. в данном случае процесс растворения экзотермический, он сопровождается выделением теплоты (ΔHраств
<0). По принципу Ле Шателье равновесие в данном случае смещается влево, и концентрация газа в растворе уменьшается. Поэтому, например, на стенках стакана воды, принесенного с улицы в теплое помещение, появляются пузырьки воздуха, из-за уменьшения растворимости газа (воздуха) при повышении температуры.
Если говорить о растворимости газов в жидкостях, то необходимо отметить, что она различна. Некоторые газы очень мало растворимы в воде (азот, водород), расворимость других газов – очень велика. Значительную растворимость аммиака в воде можно объяснить его химическим взаимодействием с водой.
На растворимость газов сильно влияет давление. Зависимость растворимости газов от давления выражается законами Дж. Генри и Дж. Дальтона.
1.
· Масса газа, растворяющегося в данном объеме жидкости, пропорциональна давлению, которое газ производит на жидкость.
По другому, закон Генри можно сформулировать и так:
· Растворимость газа при постоянной температуре прямопропорциональна его порциальному давлению над раствором.
 Х=КР Х=КР
Здесь Х– молярная доля растворенного вещества в насыщенном растворе. К– коэффициент пропорциональности (константа Генри), Р– парциальное давление.
2. Объем газа, растворяющегося в данном объеме жидкости не зависит от давления.
(Например, при 200
С и 1атм в 100г воды растворяется 0,17г диоксида углерода. При увеличении давления в два раза масса растворяющегося газа тоже увеличивается в два раза и будет равной 0,34г. В тоже время объем газа не изменяется).
3. При растворении смеси газов растворимость каждой составной части пропорциональна своему порциальному давлению (той части общего давления в газовой смеси, которая обусловлена данным газом).
Следует отметить, что эти законы имеют ограничения. Они справедливы только для сравнительно разбавленных растворов, невысоких давлений и отсутствия химического взаимодействия между молекулами газа и растворителя. Газы, вступающие в химическое взаимодействие с водой, этим законам не подчиняются. Но, в принципе, растворимость газа при нагревании обычно уменьшается, а с увеличением давления повышается.
в). Растворимость жидкостей в жидкостях.
Интервал растворимости жидкостей в жидких растворителях значительный. Одни из них смешиваются в любых отношениях (например вода и спирт). Вторые растворяются друг в друге до определенного предела. (Так, если смешать эфир с водой, то образуется два слоя: верхний слой представляет собой насыщенный раствор воды в эфире, а нижний – насыщенный раствор эфира в воде. С увеличением температуры частичная растворимость двух жидкостей друг в друге, как правило, возрастает.
3. Присутствие посторонних веществ.
Как правило посторонние вещества своим присутствием в растворе уменьшают растворимость данного вещества. Уменьшение растворимости вещества в присутствии солей обычно называют ”высаливанием”. Так же происходит уменьшение растворимости малорастворимых электролитов, если ввести в их насыщенный раствор одноименные ионы.
4. На скорость процесса растворения влияют степень диффузии растворяемого вещества в среду растворителя (необходимо применение перемешивания) и дисперстность (степень измельчения) твердого вещества.
11.3. Способы выражения концентраци растворов.
Важной характеристикой любого раствора является относительное содержание в нем растворенного вещества и растворителя, которое называется концентрацией. Количественно концентрация может выражаться разными способами: отношением масс, объемов, числа молей, отношением массы к объему и, наоборот, числа молей к массе или объему и т.д. Одни способы выражения концентрации относятся к так называемым весовым способам, а другие –к объемным. На практике используют более десятка (точнее тринадцать) способов выражения концентрации.
1. Массовая доля: Nm
=m/m+m0
(отношение массы растворенного вещества
“m” к массе раствора, т.е. сумме масс ве-
щества и растворителя (m0
)).
2. Мольная доля: Nn
=n/n+n0
(отношение числа молей растворенного ве-
щества “n” к сумме числа молей вещества и растворителя).
3. Объемная доля: Nv
=v/v+v0
(отношение объема растворенного вещества
к сумме объемов вещества и растворителя).
4. Массовый процент: m/m+m0
*100, %
(обычно этот способ называют процентной концентрацией).
5. Мольный процент: n/n+n0
*100, %.
6. Объемный процент: v/v+v0
*100, %.
7. Массовое отношение: m/m0
(отношение массы вещества к массе раство-
рителя).
8. Объемное отношение: v/v0
(отношение объема растворенного вещества
к объему растворителя).
9. Мольное отношение: n/n0
(отношение числа молей растворенного ве-
щества к числу молей растворителя).
10. Молярная концентрация (или молярность).
Определяется отношением числа молей растворенного вещества к объему раствора, выраженному в литрах. Физический смысл молярной концентрации заключается в том, что она указывает на число молей вещества содержащегося в 1литре его раствора. Обозначают М или См.
11. Нормальная концентрация (или нормальность).
Определяется отношением числа эквивалентов растворенного вещества к объему раствора, выраженному в литрах. Физический смысл нормальной концентрации заключается в том, что она указывает на число эквивалентов растворенного вещества, содержащегося в 1литре раствора. Обозначают Н или Сн.
12. Моляльная концентрация (моляльность).
Определяется отношением числа молей растворенного вещества к массе растворителя, выражается в килограммах. Физический смысл заключается в том, что она показывает, сколько молей вещества растворено в 1кг (1000г) растворителя. Обозначают m или Сm. Моляльность можно расчитать по следующей формуле: m=1000*a/Ma*b (где а –масса растворенного вещества в граммах, Ма –молекулярная масса вещества, b –масса растворителя).
13. Титр. (Т.) указывает на массу в граммах растворенного вещества, содержащуюся в одном миллилитре (см3
) раствора.
Применение того или иного способа выражения концентрации зависит от решения конкретных практических задач.
|



